首页 > 医疗资讯/ 正文
导读
胰腺导管腺癌(pancreatic ductal denocarcinoma, PDAC)是最致命的癌症之一,其五年生存率仅为13%。PDAC缺乏有效治疗的主要原因是肿瘤微环境间质压高、血管稀少和营养缺失。为适应此营养匮乏的微环境,PDAC细胞依赖溶酶体途径,通过表达高亲和力营养转运蛋白、大量吞噬以及与其他细胞相互作用等手段来获取非常规的营养物质。研究报道,靶向自噬和溶酶体依赖性途径能破坏PDAC代谢,但是缺乏有效且具体的靶点。
脂质激酶PIKfyve能催化磷脂酰肌醇3,5-二磷酸(PtdIns (3,5) P2)和磷脂酰肌醇5-磷酸(PtdIns5P),在溶酶体途径中发挥重要的调控作用。目前,两种PIKfyve抑制剂apilimod和ESK981已通过1期临床试验,提示靶向PIKfyve具有巨大的转化潜力。
近日,来自美国密歇根大学的研究团队在Nature上发表了题为“Targeting PIKfyve-driven lipid metabolism in pancreatic cancer”的研究报告。研究发现,抑制PIKfyve会迫使PDAC依赖脂质从头合成以获取能量,从而遏制PDAC发展,而联合抑制KRAS-MAPK(kirsten rat sarcoma viral oncogene homolog,mitogen-activated protein kinase)通路能够阻断脂质从头合成,有效清除PDAC。这种双重靶向脂质稳态的策略有望成为PDAC极具前景且可快速转化的新疗法。

表型:PIKfyve抑制剂显著抑制胰腺癌进展
为探究PIKfyve在胰腺癌发展中的作用,本研究:
-
利用RNA原位杂交自发技术发现,癌变组织部位中PIKfyve呈高表达。
-
通过构建胰腺腺泡细胞(Ptf1a-Cre)特异性敲除PIKfyve的转基因小鼠,发现在健康小鼠中敲除PIKfyve不影响胰腺的发育和功能。然而,在KPC模型中敲除PIKfyve或使用PIKfyve抑制剂(ESK981),均可显著延长小鼠生存时间,并抑制肿瘤进展。
-
在皮下同种异体移植瘤模型和异种移植瘤模型中,ESK981均表现出良好的抗肿瘤活性,显著抑制了肿瘤生长,并降低了肿瘤负荷。
上述结果表明:胰腺在健康状态下不需要PIKfyve,但在PDAC发病时,抑制PIKfyve能有效抑制肿瘤进展。
机制:抑制PIKfyve降低溶酶体自噬功能并诱导脂肪酸从头合成
(1)PIKfyve调控PDAC进展的分子机制
本研究分别采用基因敲除PIKfyve、使用PIKfyve抑制剂(apilimod、ESK981)或靶向降解剂(PIK5-33d)处理不同胰腺癌细胞,发现细胞出现自噬通量降低,增殖活性减弱以及细胞空泡化。虽然自噬抑制剂氯喹也能产生类似效果,但PIKfyve抑制显示出更强的杀伤活性。
基于溶酶体(包括自噬)在维持铁稳态和促进线粒体呼吸中的作用,研究团队进一步探究PIKfyve抑制是否通过类似的机制抑制细胞生长。结果发现,PIKfyve抑制并未降低氧耗率,其抗增殖作用也无法被铁补充所逆转,表明PIKfyve抑制不依赖铁稳态或线粒体呼吸来发挥其抑制肿瘤的作用。
(2)评估PIKfyve在代谢中的调控作用
本研究对MIA PaCa-2细胞进行了两轮平行CRISPR筛选(高/低剂量PIKfyve抑制),聚焦于代谢相关基因。结果显示,PIKfyve抑制导致脂质合成通路基因的显著耗竭:高剂量抑制靶向胆固醇合成、脂肪酸从头合成与延伸、鞘脂合成和线粒体代谢;低剂量抑制靶向脂肪酸合成与延伸,同时编码脂质β-氧化第一步限速酶ACOX1的sgRNA在两种剂量筛选中均被强烈富集。
本研究进一步使用CRISPRi技术或小分子化合物特异性抑制脂质合成通路关键蛋白的活性,发现均能增加细胞对PIKfyve抑制剂的敏感性。综上表明,PIKfyve抑制后,细胞自噬通量降低并诱导细胞通过增强脂质分解(β-氧化)来补偿脂质合成阻断引发的代谢危机。
(3)PIKfyve抑制后PDAC细胞的组学分析
-
转录组分析显示,上调基因主要富集于胆固醇稳态、mTORC1信号通路以及脂肪酸代谢等代谢相关通路。其中,典型SREBP1靶基因和SREBP1表达升高,而SREBP阻断剂可部分逆转上述效应,这提示SREBP在PIKfyve调控脂质代谢中发挥重要作用。
-
代谢组分析显示,抑制PIKfyve后,细胞呈现与转录组相似的代谢谱变化。
-
靶向脂质组分析显示,PIKfyve抑制诱导脂质组成发生显著改变,且这些变化部分依赖于SREBP1活性。
鉴于柠檬酸转运体SLC25A1在CRISPR筛选中也被鉴定为关键靶点,本研究提出假设:糖酵解代谢物被用于生成柠檬酸,后者转而分流至从头脂质合成,并且通过U-¹³C₆葡萄糖稳定同位素示踪实验证实该假设。这些数据表明,PIKfyve抑制迫使PDAC细胞将葡萄糖来源的碳流转向新脂质(尤其是鞘脂)的合成。
(4)PIKfyve抑制导致溶酶体膜胆固醇积累,激活SREBP1信号
研究发现,PIKfyve抑制并未干扰AMPK信号通路,且其对SREBP的激活作用不依赖于AMPK。同时,自噬启动因子ULK1抑制、其他溶酶体抑制剂及溶酶体胆固醇转运蛋白NPC1抑制剂,均能激活SREBP1信号。PIKfyve抑制导致溶酶体空泡化和溶酶体膜胆固醇异常积累,而U18666A仅引起胆固醇滞留,不诱发空泡化。值得注意的是,溶酶体膜胆固醇积累与SREBP激活在4小时即出现,且外源补充固醇可逆转PIKfyve抑制诱导的SREBP激活。
综上表明,PIKfyve抑制破坏脂质稳态的机制独立于AMPK信号与自噬流,其通过引起溶酶体空泡化和功能障碍,导致脂质(尤其是胆固醇)滞留在溶酶体膜,进而迫使细胞依赖脂质从头合成以维持生存。
治疗:双重抑制PIKfyve与KRAS-MAPK通路实现协同抗癌
既往研究报道,KRAS-MAPK信号转导驱动脂肪酸从头合成。与之一致,本研究发现抑制KRAS-MAPK信号转导可抑制脂肪酸从头合成途径。基于此,本研究进一步探究了联合抑制PIKfyve和KRAS-MAPK通路是否能协同阻断脂质合成,进而抑制肿瘤进展。实验结果显示:
-
单用PIKfyve抑制剂会促进脂肪酸从头合成关键酶(FASN/ACC1)表达上调,但联合KRAS失活后,该上调效应被显著抑制。
-
在体外细胞实验中,PIKfyve抑制剂(apilimod/ESK981)与KRAS-MAPK抑制剂(MRTX1133/Trametinib)任意组合均显示出强协同效应,显著降低PDAC细胞的增殖活力。
-
在动物模型中,通过三类递进模型(免疫健全的同源原位模型→人源PDX模型→自发成瘤KPC模型)进一步发现了双重抑制对肿瘤的杀伤作用,具体而言:几乎完全清除肿瘤,中位生存期延长超过5倍,消除肿瘤效果显著且优于单药治疗。
以上实验结果充分验证了治疗策略的转化潜力,为后续临床试验奠定基石。
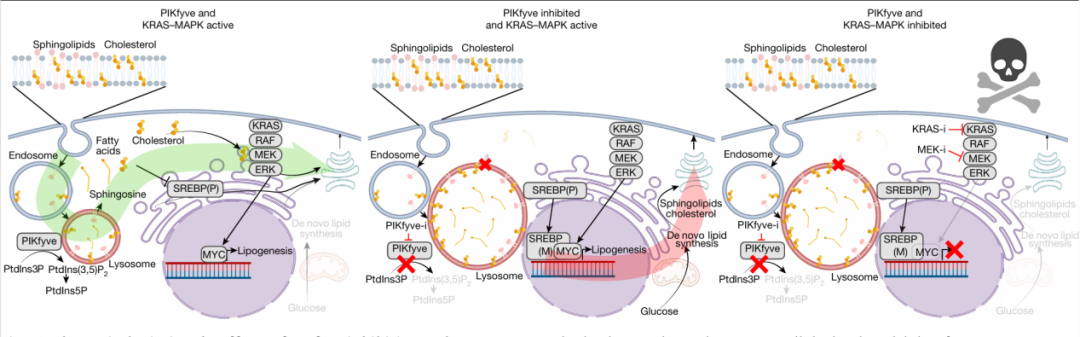
图1:单PIKfyve抑制作用以及协同KRAS-MAPK双重抑制作用
图源:Nature原文
研究总结
本研究证实PIKfyve是破坏PDAC溶酶体功能的治疗靶点,并详细阐明了其作用机制,同时发现联合抑制PIKfyve与KRAS-MAPK通路可显著消除PDAC肿瘤负荷。
PIKfyve是维持溶酶体依赖性脂质稳态的必要调节因子,而KRAS-MAPK信号驱动PDAC细胞的从头脂质合成;PIKfyve抑制导致溶酶体脂质代谢紊乱,迫使PDAC细胞上调并依赖从头脂质合成;双重抑制PIKfyve与KRAS-MAPK使PDAC细胞因无法获取功能性脂质而陷入代谢危机。
鉴于突变型KRAS抑制剂、泛KRAS抑制剂、MAPK通路抑制剂及PIKfyve抑制剂ESK981的快速发展,PIKfyve与KRAS-MAPK抑制剂的联合使用,将有望成为治疗PDAC极具前景且具备快速转化潜力的治疗策略。
引证本文
Cheng, C., Hu, J., Mannan, R. et al. Targeting PIKfyve-driven lipid metabolism in pancreatic cancer. Nature (2025).
https://doi.org/10.1038/s41586-025-08917-z
猜你喜欢
- Nature:疫情期间儿童近视激增:危机与应对策略
- 眼霜晚上睡觉可以用吗 晚上用好还是早上用好
- 吉林大学中日联谊医院流式细胞仪采购项目中标公告
- JAMA Otolaryngol Head Neck Surg:人工耳蜗植入与青少年的教育和生活质量结果评估
- Front Immunol:男男性行为人群HIV早期检测中HIV自我检测和机构检测方法的比较
- 25省份明确社会抚养费标准 你的家乡超生罚多少
- 感染性休克中的升压药物
- 指南解读 | 晚期肝细胞癌的系统治疗:2024 ASCO指南更新临床视角
- 小欧探案记|糖尿病合并多重并发症患者TG飙升至59.93mmol/L!该如何破解血脂管理困局?
- 5部门发布指导意见 促进医养结合服务高质量发展
- 搜索
-
- 1000℃Nutrients:真实世界数据,纤维肌痛患者的饮食与运动自适应规律
- 1000℃D-二聚体升高诊治与管理专家共识(2026)
- 1000℃专家论坛|文良志:门静脉血栓的诊断和治疗
- 1000℃首例儿童NF2驱动型胸膜间皮瘤,多方法学检测锁定NF2双等位基因失活和14/22号染色体缺失,提示与成人胸膜间皮瘤不同
- 1000℃打破误区:干扰素追求CHB功能性治愈,HBsAg为何“不降反增”?
- 1000℃迷惑性极强的肝内病灶!影像表现疑点重重,最终病理竟查出两种不同肝脏恶性肿瘤
- 1000℃指南共识|原发性肝癌分子靶向药物相关蛋白尿中西医结合诊疗专家共识
- 1000℃Diabetologia:意大利北部社区 1~100 岁人群胰岛自身抗体与乳糜泻 TGA-IgA 的年龄分布及检测方法学验证
- 精J Child Psychol Psychiatry:12种罕见神经发育障碍儿童沟通能力谱系
- 精研究发现:爱吃辣的人,心血管病和癌症死亡风险都会显著降低
- 精Nursing in Critical Care:别再指责护士了!ICU 里被遗漏的护理,根源在系统而非个人
- 精Acta Obstet Gynecol Scand:罕见病女性的妊娠并发症与母婴结局,一项单中心434种罕见病的回顾性队列研究
- 精【爱儿小醉】儿科患者术前对流层臭氧暴露与围手术期呼吸系统不良事件之间的关系:一项单中心回顾性队列研究
- 精eBioMedicine:牙龈下微生物组与脑健康存在连续关联梯度,牙周炎或成认知衰退可干预靶点
- 精军事医学研究院《自然·通讯》:自适应IrPtCu纳米酶水凝胶实现耐药菌感染伤口序贯治疗
- 精能够逆转萎缩性胃炎的两个中成药,该怎么选择?
- 荐同时性多发性原发性肺癌,左右病灶分别为EGFR和ALK阳性,考虑淋巴结肿大仅局限左肺门及血浆EGFR阳性,采用奥希替尼联合化疗
- 荐40岁女性同时罹患卵巢支持细胞-间质细胞瘤和透明细胞乳头状肾肿瘤,WES等基因检测竟为阴性
- 荐椎管内麻醉使用止血药突发气道痉挛的抢救流程解析
- 荐女子肝区无任何不适,影像提示复杂囊性病变,层层鉴别后锁定罕见胆管源性囊性肿瘤
- 荐“绘”真报告 | 病理考虑为中枢神经细胞瘤,检出脑室外神经细胞瘤的特征性变异FGFR1-TACC1融合,辅助鉴别诊断
- 荐8岁女童出现男性化症状,竟是形似「性索-间质肿瘤」的卵巢「无性细胞瘤」所致,少见KRAS/CDK4共扩增或与侵袭性有关
- 荐17例病例分析揭示常见于中年人的色素性室管膜瘤临床特征与预后,分子检测可助力临床精准诊疗
- 荐Lancet Oncol:结直肠癌腹膜转移,围手术期化疗并非必选项
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)