首页 > 医疗资讯/ 正文
心肌肥大是导致心力衰竭的重要病理变化。最近的研究表明,线粒体相关内质网膜(MAM)在这一病理过程中起着关键作用。然而,分子机制尚不清楚。
2025年6月9日,浙江大学胡新央、Zhu Wei共同通讯在Circulation在线发表题为“FMO2 Prevents Pathological Cardiac Hypertrophy by Maintaining the ER-Mitochondria Association Through Interaction With IP3R2-Grp75-VDAC1”的研究论文,该研究表明FMO2通过与IP3R2-Grp75-VDAC1相互作用维持ER线粒体关联,从而预防病理性心肌肥大。
在这里,研究人员检测到MAM结构中一个意想不到的成分,即FMO2,一种内质网驻留蛋白。病理性心肌肥大期间,FMO2水平降低。FMO2的缺失和过表达分别会恶化或阻止体内病理性心力衰竭的进展。进一步证明,FMO2定位于MAM结构,在那里它与肌醇1,4,5-三磷酸2型受体(IP3R2)结合,作为IP3R2-Grp75(葡萄糖调节蛋白75)-VDAC1(电压依赖性阴离子通道蛋白1)复合物的一个组成部分,维持内质网-线粒体接触,并调节线粒体Ca2+信号传导,用于生物能量学。研究发现,一种增强内质网-线粒体接触的合成肽促进了Ca2+的转移,并预防了病理性心肌肥大。该研究结果揭示了FMO2在心肌肥大中的关键作用,FMO2在维持MAM结构和功能方面起着关键作用,这可能代表了心肌肥大和心力衰竭的新机制和治疗靶点。
 心力衰竭作为各种心血管疾病的终末期,已被公认为全球死亡率和发病率的主要原因,这引发了人们对该研究领域的极大兴趣。从适应性心肌肥厚到不适应性心肌肥厚的转变与心力衰竭的进展密切相关,是心脏死亡的独立危险因素。尽管人们对不适应性心肌肥大的研究做出了巨大努力,但心肌肥大的潜在机制尚未完全了解。
心力衰竭作为各种心血管疾病的终末期,已被公认为全球死亡率和发病率的主要原因,这引发了人们对该研究领域的极大兴趣。从适应性心肌肥厚到不适应性心肌肥厚的转变与心力衰竭的进展密切相关,是心脏死亡的独立危险因素。尽管人们对不适应性心肌肥大的研究做出了巨大努力,但心肌肥大的潜在机制尚未完全了解。
最近的研究强调了细胞器相互作用在维持细胞稳态和调节各种病理过程中的关键作用,包括病理性心肌肥大。内质网(ER)似乎是细胞器交叉相互作用中最活跃的参与者,特别是与线粒体的相互作用,这已成为细胞生物学领域的一个热门话题。线粒体相关ER膜(MAM)是由特定的束缚和间隔蛋白形成的动态微区,在Ca2+信号传导、脂质转运和代谢中起着至关重要的作用。MAM在从适应性心肌肥大向不适应性心肌肥大的过渡过程中发生了变化,导致心力衰竭进展。然而,MAM的结构和功能如何受到调节并导致心力衰竭的进展(即病理性心肌肥大)尚未完全阐明。
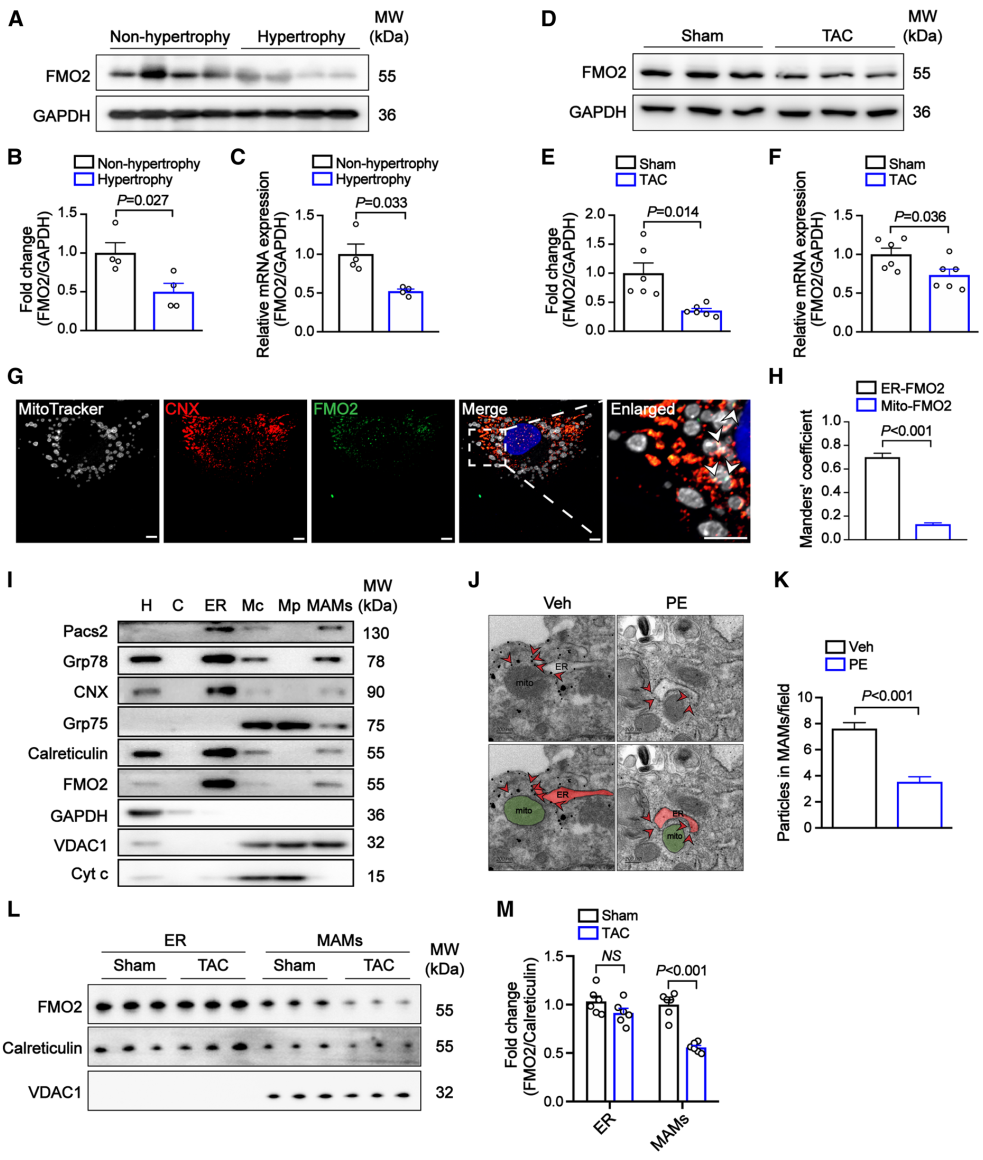
FMO2定位于MAM,在肥大的心脏中其表达减少(图源自Circulation)
有趣的是,最近的研究表明,在MAM复合物中也检测到FMO(含黄素单加氧酶)家族蛋白。FMO蛋白是一组微粒体酶,其跨膜结构域主要与ER结合。FMO2是心脏中的主要亚型,已被证明在长寿和病理纤维化中发挥作用,但其在MAM中的作用尚未得到探索。
在这项研究中,对人类肥大心脏组织的大量RNA测序分析和小鼠肥大心脏组织中MAM靶向蛋白质组学都揭示了MAM中FMO2表达的改变。进一步的实验表明,FMO2与肌醇1,4,5-三磷酸2型受体(IP3R2)结合,参与形成IP3R2/葡萄糖调节蛋白75(Grp75)/电压依赖性阴离子通道蛋白1(VDAC1)复合物,这是MAM的关键结构成分。FMO2表达的变化通过改变MAM的形成来影响ER向线粒体释放Ca2+,从而主要通过调节线粒体Ca2+水平来干扰线粒体生物能量学。因此,FMO2在心肌肥大的发病机制中起着至关重要的作用,其过表达可以预防适应不良的心肌肥大。值得注意的是,通过合成连接肽连接ER和线粒体可以增加线粒体内的Ca2+水平,并防止病理性心肌肥大的进展。总的来说,该研究不仅发现了MAMs复合物的一个新成分,还为病理性心肌肥大提供了一个新的治疗靶点。
参考信息:https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.124.072661
- 上一篇: 河北关于公示公布部分药品挂网的通知
- 下一篇:穿刺肱动脉行介入治疗的操作技巧
- 搜索
-
- 1000℃西南大学/Biomaterials:可见光响应阳离子化光动力纳米颗粒整合丝素蛋白纳米纤维基质通过增强ROS利用促进感染创面愈合
- 1000℃空军军医大学Biomaterials:增强压电胶原各向异性支架通过电-拓扑协同信号促进骨骼肌再生
- 1000℃南方医科大学Biomaterials:光热-压电双响应抗菌假体界面调控施万细胞表型转化促进感染性骨缺损修复
- 1000℃南京大学Biomaterials:生物硅纳米笼可注射水凝胶捕获内源性TGF-β1用于原位透明软骨再生
- 1000℃Nat Genet | 刘斯洋/魏凤香/张铨富等绘制人类妊娠动态遗传图谱,揭示妊娠表型的特异性遗传效应
- 1000℃华南理工大学,最新Biomaterials:3D打印仿生海绵体联合间充质干细胞通过免疫微环境重塑修复雄性猪勃起功能
- 1000℃内蒙古大学AM:一种协同工程的蛋白质水凝胶,整合拓扑强化和金属离子协调,用于糖尿病伤口修复
- 1000℃同济大学附属东方医院Biomaterials:钒单原子催化剂通过GPR110依赖性巨噬细胞极化介导脓毒症肠损伤治疗
- 精J Child Psychol Psychiatry:12种罕见神经发育障碍儿童沟通能力谱系
- 精研究发现:爱吃辣的人,心血管病和癌症死亡风险都会显著降低
- 精Nursing in Critical Care:别再指责护士了!ICU 里被遗漏的护理,根源在系统而非个人
- 精Acta Obstet Gynecol Scand:罕见病女性的妊娠并发症与母婴结局,一项单中心434种罕见病的回顾性队列研究
- 精【爱儿小醉】儿科患者术前对流层臭氧暴露与围手术期呼吸系统不良事件之间的关系:一项单中心回顾性队列研究
- 精eBioMedicine:牙龈下微生物组与脑健康存在连续关联梯度,牙周炎或成认知衰退可干预靶点
- 精军事医学研究院《自然·通讯》:自适应IrPtCu纳米酶水凝胶实现耐药菌感染伤口序贯治疗
- 精能够逆转萎缩性胃炎的两个中成药,该怎么选择?
- 荐同时性多发性原发性肺癌,左右病灶分别为EGFR和ALK阳性,考虑淋巴结肿大仅局限左肺门及血浆EGFR阳性,采用奥希替尼联合化疗
- 荐40岁女性同时罹患卵巢支持细胞-间质细胞瘤和透明细胞乳头状肾肿瘤,WES等基因检测竟为阴性
- 荐椎管内麻醉使用止血药突发气道痉挛的抢救流程解析
- 荐女子肝区无任何不适,影像提示复杂囊性病变,层层鉴别后锁定罕见胆管源性囊性肿瘤
- 荐“绘”真报告 | 病理考虑为中枢神经细胞瘤,检出脑室外神经细胞瘤的特征性变异FGFR1-TACC1融合,辅助鉴别诊断
- 荐8岁女童出现男性化症状,竟是形似「性索-间质肿瘤」的卵巢「无性细胞瘤」所致,少见KRAS/CDK4共扩增或与侵袭性有关
- 荐17例病例分析揭示常见于中年人的色素性室管膜瘤临床特征与预后,分子检测可助力临床精准诊疗
- 荐Lancet Oncol:结直肠癌腹膜转移,围手术期化疗并非必选项
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)