首页 > 医疗资讯/ 正文
导读
自噬关键因子(autophagy related 14, ATG14)除了参与自噬的起始与自噬体-溶酶体融合过程外,其在调节肝细胞活力中的作用尚不清楚。近日,来自美国印第安纳大学Charlie Dong教授及团队在eGastroenterology发表了题为“Autophagy related 14 protects against liver injury by inhibiting multiple cell death pathways”的最新成果。研究发现,ATG14缺失会引发凋亡、坏死性凋亡和焦亡等多种细胞死亡通路并行激活,导致明显的肝损伤、炎症与纤维化加重,提示ATG14对维持肝脏稳态具有重要的保护作用。

研究设计与方法
1. 构建小鼠模型
本研究采用基因工程小鼠模型,系统探讨ATG14在肝脏中的生理功能。通过AAV8-TBG-Cre系统成功构建Atg14肝脏特异性敲除(Atg14 HepKO)小鼠模型。此外,部分实验小鼠采用Alb-Cre系统实现肝脏特异性敲除,用于透射电子显微镜分析肝细胞形态学变化。
2. 饮食干预
研究者使用模拟高脂饮食的西方饮食(Western diet, WD)方案,对实验小鼠连续喂养4周,以诱导脂肪肝样代谢性肝损伤。此种饲料的配方中含有40%脂肪、40%糖类(其中20%为果糖)以及1%胆固醇,用于系统评估ATG14缺失在不同代谢状态下对肝脏功能的影响。
3. 实验检测
研究团队采用多种手段系统评估肝脏结构和功能的变化:
-
血清生化检测:测定丙氨酸氨基转移酶(alanine aminotransferase, ALT)、天冬氨酸氨基转移酶(aspartate aminotransferase, AST) 及血脂等指标,用于评估肝功能和代谢状态;
-
组织学染色(HE、Sirius Red、Trichrome):观察肝细胞损伤、炎症浸润与纤维化程度;
-
免疫印迹:验证关键蛋白的表达水平;
-
免疫组化与免疫荧光:定位和检测特定蛋白的表达和分布;
-
透射电子显微镜:观察细胞亚结构变化,重点评估自噬体与线粒体形态;
-
RNA测序:分析基因表达的整体变化。
-
TUNEL染色(transferase dUTP nick end labeling):检测细胞凋亡情况。
主要发现:从表型到机制,一环扣一环
1. 普通饮食条件下即出现肝肥大和肝损伤
-
自噬受损证据:ATG14表达下降超过90%,伴随p62/SQSTM1与LC3-I水平显著升高(见图1A),提示肝细胞自噬显著受损。
-
肝损伤表现:ALT、AST明显升高(见图1B),最突出的表型为严重肝肥大(肝重/体重比显著增加;见图1C–M)。
-
肝细胞特异性表型与再生:HNF4α表达下降、增殖标志物Ki-67上调(见图1M–N),提示肝细胞损伤与修复并存。
-
炎症与纤维化启动:肝组织中F4/80阳性巨噬细胞浸润增加;Sirius Red/Trichrome与COL1免疫信号均明显增强,炎症与纤维化信号通路显著激活,相关蛋白(iNOS、TLR4、p-RELA)上调;炎性因子Tnf、Il6、Il1b、Ccl2、Ccl5转录升高,而抗炎因子Il10下调(见图1O–S)。
要点:即使在无代谢应激条件下,ATG14缺失已可导致显著肝肥大与肝损伤,并伴随炎症和纤维化反应的激活。
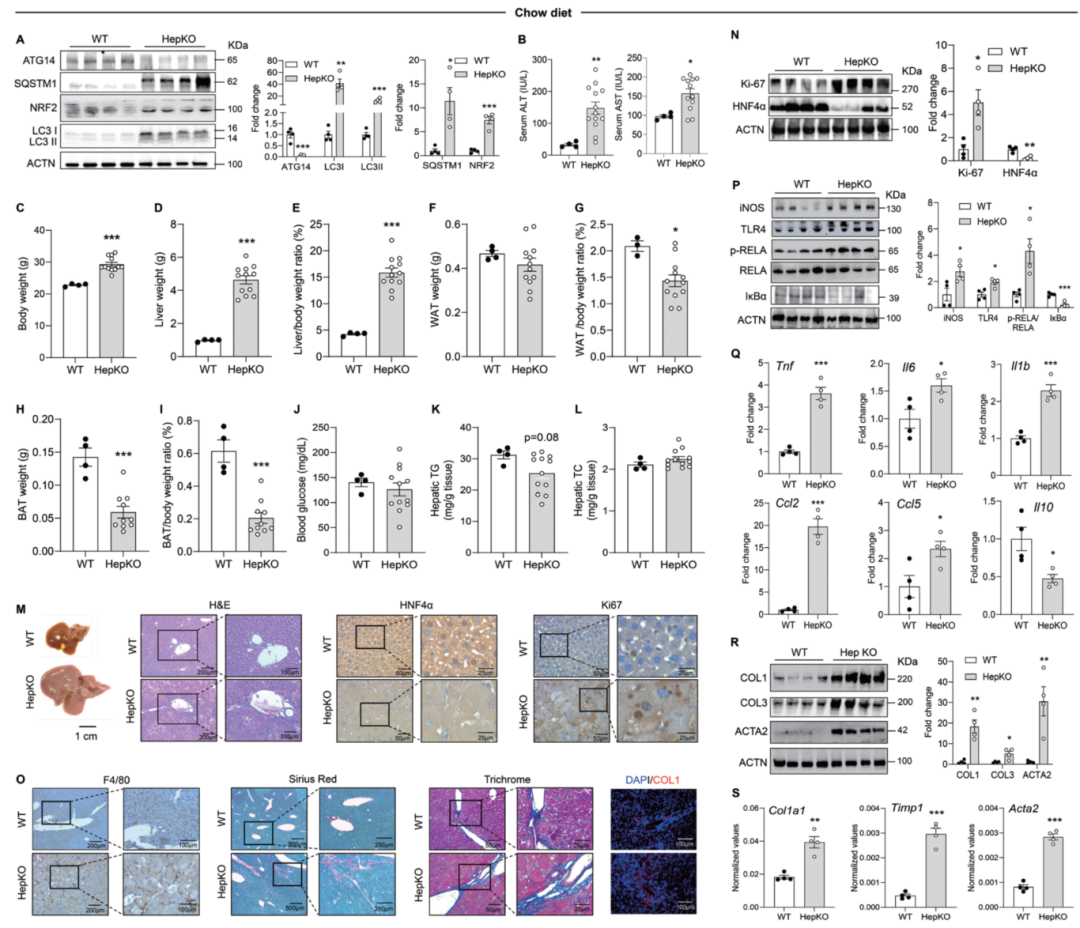
图1:Atg14 HepKO小鼠在普通饮食条件下表现出自噬受损、肝肥大及伴随的肝损伤、炎症与纤维化表型。
2. 西方饮食4周:炎症、纤维化与组织破坏整体加剧
-
自噬缺陷与肝损伤加重:在西方饮食喂养4周后(见图2A),p62与LC3-I/II水平持续累积;ALT、AST显著升高(见图2B–D);H&E染色显示肝实质结构严重破坏,同时HNF4α明显下降、Ki-67表达升高(见图2E–G)。
-
炎症-纤维化耦合加剧:肝组织中F4/80阳性细胞浸润显著增加;血清及肝脏TNF-α水平均升高;羟脯氨酸含量增多;NF-κB信号通路被激活(p-RELA上调,IκBα下调);纤维化相关信号轴显著增强:TGFBR1、p-SMAD3,PDGFRB,COL1/3与TIMP1均呈升高趋势。
-
脂质代谢特征:血清甘油三酯中度升高,但肝内甘油三酯反而含量下降(见图2M–P),与此前报道的Atg5、Atg7、FIP200 HepKO小鼠表型一致。
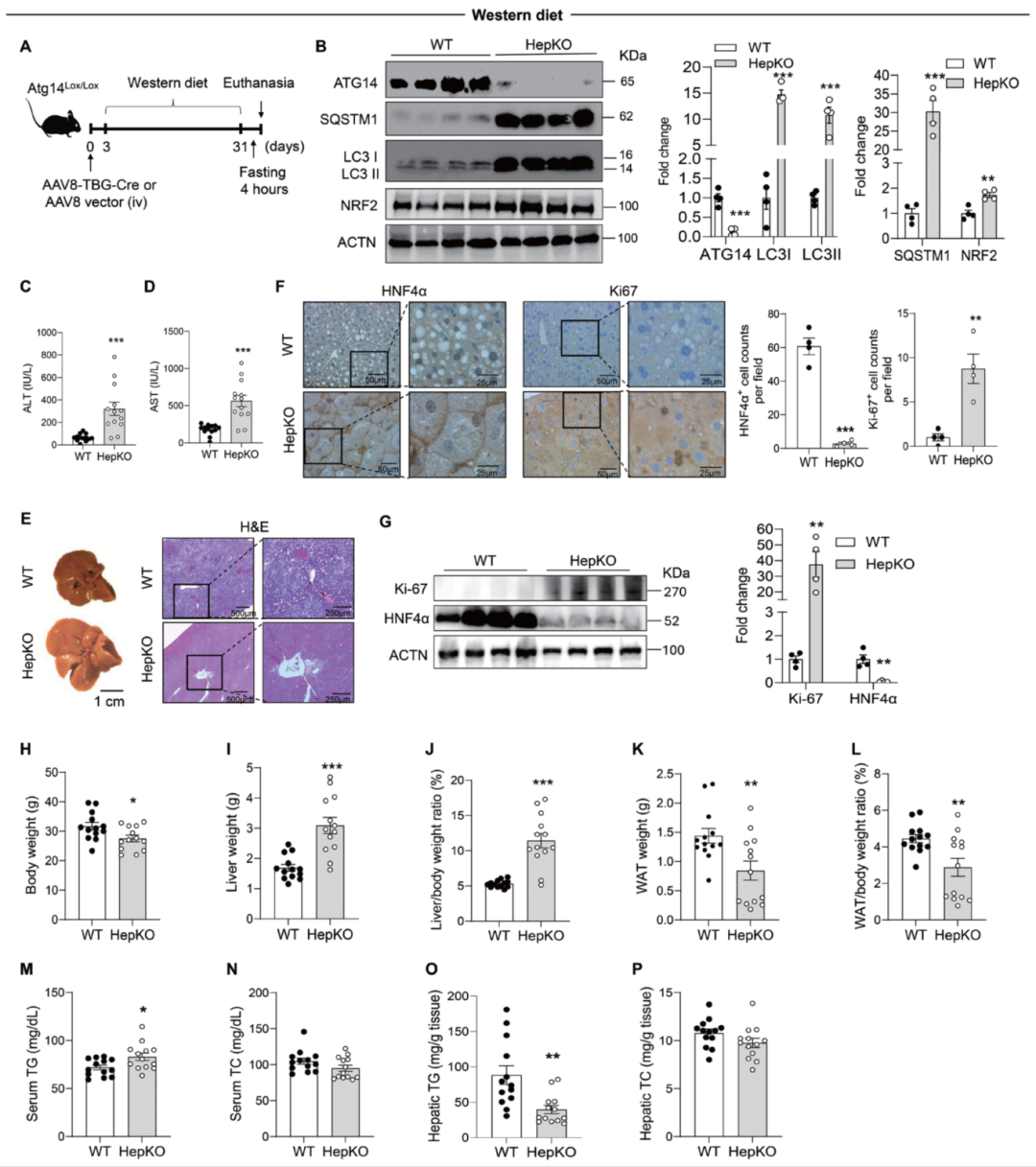
图2:Atg14 HepKO小鼠在西方饮食喂养条件下表现出肝功能损伤、细胞增殖改变及代谢紊乱。
3. 线粒体与氧化还原:能量工厂“坍塌”
-
应激与氧化还原失衡:在西方饮食喂养条件下,4-HNE与MDA蛋白加成增加;谷胱甘肽(glutathiones, GSH)及其还原比例GSH/(GSH+GSSG(oxidised glutathiones))下降。
-
呼吸链与超微结构异常:线粒体呼吸链复合体I、III、V的蛋白水平显著降低;透射电子显微镜下可见线粒体形态大多不规则、延长、嵴结构紊乱,并伴有异常多层膜样结构。
要点:ATG14缺失与线粒体结构破坏及氧化还原失衡密切相关,放大了活性氧(reactive oxygen species, ROS)生成与细胞死亡通路的激活。
4. 多通路细胞死亡被“坐实”,PANoptosis关键分子同步上调
-
多通路细胞死亡证据确立:TUNEL染色显示,在西方饮食喂养条件下,ATG14缺失小鼠肝组织中阳性细胞数量显著增加。
-
三大细胞死亡通路激活:凋亡标志物cleaved-CASP3升高、坏死性调往标志p-MLKL上调、焦亡相关蛋白AIM2、NLRP3、cleaved-GSDMD、cleaved-CASP1水平均显著增加。
-
PANoptosis组件同步上调:关键调控分子ZBP1、RIPK1、IRF1、p-STAT1以及cleaved-CASP8均同步上调。
要点:既往研究已证实TNF-α和IFN-γ可通过JAK-STAT1-IRF1-iNOS轴诱导PANoptosis。本研究发现,在ATG14缺失的背景下,该信号轴被显著激活。
5. 细胞定位:焦亡主要发生于肝细胞,炎症小体活化集中于巨噬细胞
免疫荧光共定位显示:GSDMD/GSDME主要在肝细胞中升高;而NLRP3主要在肝巨噬细胞中升高。这一结果不仅与“肝细胞特异性敲除”模型的设计相符,也为后续观察到的局灶性炎症与纤维化放大效应提供了合理的解释。
讨论精要:几个“看点”与“难点”
1. 为何肝内甘油三酯不升反降?
在无论是普通饮食还是西方饮食喂养条件下,Atg14 HepKO小鼠的肝内甘油三酯水平均呈下降趋势。这与Atg5、Atg7、FIP200 HepKO小鼠研究的观察一致。作者提出三种可能机制加以解释:
-
自噬缺失导致NCoR1积累,从而抑制LXRα介导的脂质生成相关基因转录;
-
自噬缺失导致NRF2持续激活,影响脂滴生成;
-
自噬系统的全面崩溃同时影响肝脏中脂滴生成与周转的多个环节,形成一种功能性“低脂滴”状态。
2. PANoptosis何以成为“并发”的分子底盘?
PANoptosis可由各种PAMPs或DAMPs引发,其发生依赖于ZBP1、AIM2、RIPK1、NLRP3、CASP8等构成的PANoptosome复合物。在ATG14缺失肝脏中,这些关键节点分子呈整体上调。与此同时,TNF-α与IFN-γ介导的JAK-STAT1-IRF1-iNOS炎症信号轴被显著激活,驱动细胞凋亡、焦亡与坏死性凋亡的并行放大,构建出一个“多通路协同的细胞死亡底盘”。
3. 肝肥大与表型改变
在Atg14 HepKO小鼠中,出现了显著的肝肥大、HNF4α下调和Ki-67上升等现象。尽管已有研究指出NRF2、YAP等通路可参与肝肥大,但本研究采用的成年期AAV-Cre特异性敲除模型有效规避了发育期的代偿干扰,从而为慢性肝病的病理演进提供了更贴近临床的模拟体系。
临床与转化启示
维持肝细胞自噬功能是抑制炎性细胞死亡级联、减轻炎症与纤维化的关键环节,构成了慢性肝病管理中的核心“底盘”机制。
ATG14-轴具备潜在的药物靶点价值,可通过维持自噬活性、调控ROS与线粒体稳态、阻断JAK-STAT1-IRF1-iNOS及NF-κB/TGF-β/PDGF等炎性信号节点来实现干预。
对于已有慢性肝病基础的患者,探索并分层实施PANoptosis抑制策略(尤其是在高促炎微环境种)或可成为一种具有前瞻性的治疗思路。
结语
ATG14缺失可引发多条细胞死亡通路并行激活,推动“肝损伤—炎症—纤维化”的恶性循环持续放大,这一发现深刻印证了“自噬稳态,即肝脏稳态”的核心观点。
引证本文
Kim H, Huang M, Wang S, Zhang Y, Li K, Liu S, et al. Autophagy related 14 protects against liver injury by inhibiting multiple cell death pathways. eGastroenterology. 2025;3:e100181.
https://doi.org/10.1136/egastro-2025-100181
- 搜索
-
- 1000℃李寰:先心病肺动脉高压能根治吗?
- 1000℃除了吃药,骨质疏松还能如何治疗?
- 1000℃抱孩子谁不会呢?保护脊柱的抱孩子姿势了解一下
- 1000℃妇科检查有哪些项目?
- 1000℃妇科检查前应做哪些准备?
- 1000℃女性莫名烦躁—不好惹的黄体期
- 1000℃会影响患者智力的癫痫病
- 1000℃治女性盆腔炎的费用是多少?
- 标签列表
-
- 星座 (702)
- 孩子 (526)
- 恋爱 (505)
- 婴儿车 (390)
- 宝宝 (328)
- 狮子座 (313)
- 金牛座 (313)
- 摩羯座 (302)
- 白羊座 (301)
- 天蝎座 (294)
- 巨蟹座 (289)
- 双子座 (289)
- 处女座 (285)
- 天秤座 (276)
- 双鱼座 (268)
- 婴儿 (265)
- 水瓶座 (260)
- 射手座 (239)
- 不完美妈妈 (173)
- 跳槽那些事儿 (168)
- baby (140)
- 女婴 (132)
- 生肖 (129)
- 女儿 (129)
- 民警 (127)
- 狮子 (105)
- NBA (101)
- 家长 (97)
- 怀孕 (95)
- 儿童 (93)
- 交警 (89)
- 孕妇 (77)
- 儿子 (75)
- Angelababy (74)
- 父母 (74)
- 幼儿园 (73)
- 医院 (69)
- 童车 (66)
- 女子 (60)
- 郑州 (58)