首页 > 健康资讯/ 正文
深度解析医学证据,lxfs.net为你支撑决策
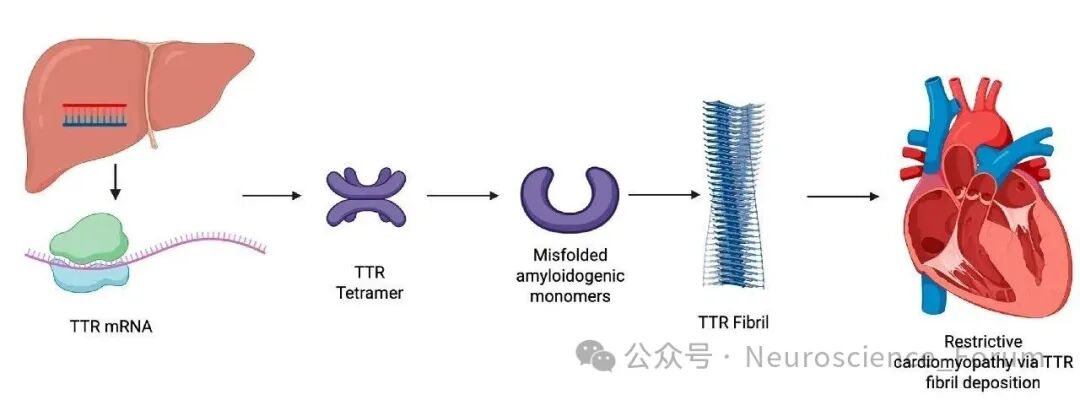
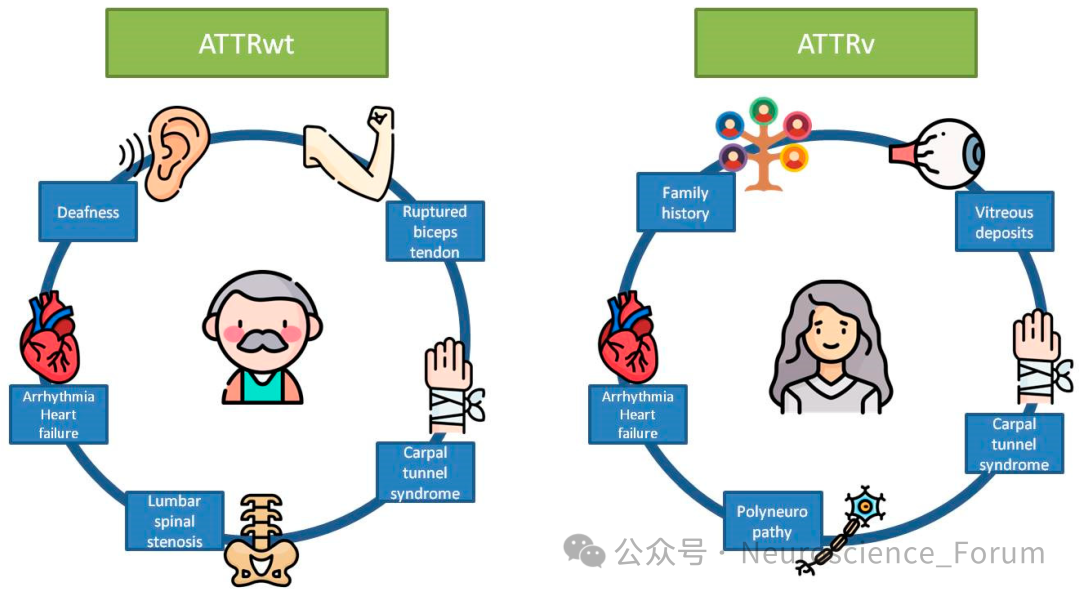
论坛导读:转甲状腺素蛋白淀粉样变(Transthyretin Amyloidosis, ATTR),是一种致死性疾病,其发病原理是由于转甲状腺素蛋白(TTR)四聚体不稳定导致单体异常折叠,形成了淀粉样沉积。这些淀粉样纤维在血液循环中逐渐沉积于人体多个组织器官,尤其是心脏和神经系统,严重影响患者的生命质量,且目前缺乏有效的治疗手段。ATTR包括治疗转甲状腺素蛋白淀粉样变性多发性神经病(Transthyretin amyloidosis polyneuropathy, ATTR-PN)和转甲状腺素蛋白心脏淀粉样变(Transthyretin amyloidosis cardiomyopathy, ATTR-CM),其中ATTR-CM又分为野生型ATTR(ATTRwt)和遗传型ATTR(hATTR)两种。ATTRwt的发病率随着年龄的增长而增加,占比接近87.5%。它主要影响老年男性,其中65岁人群当中更为多见。hATTR是由TTR基因的突变引起,患者发病年龄较早,多为50-60岁左右。
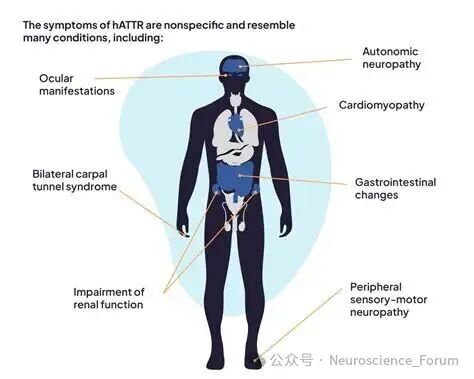
转甲状腺素蛋白淀粉样变性(Transthyretin amyloidosis,ATTR)是由于转甲状腺素蛋白(TTR)异常折叠形成淀粉样物质沉积在组织器官导致的一大类疾病。根据是否存在TTR基因突变,可分为遗传型(ATTRv)和野生型(ATTRwt)两类。ATTRv是TTR基因致病变异导致的常染色体显性遗传性多系统疾病,临床表现涉及周围神经、心脏、消化道、眼部、肾脏和软脑膜等多个系统。我国ATTRv患者的疾病特征逐渐为大家所认识,单一系统损害的患者较少,多数患者表现为周围神经、心脏等多个系统受累。
ATTRv曾被认为是一种极为罕见的疾病,全球传统估计患病人数为5 000~10000例。然而,基于基因组数据库的最新研究显示,全球人群ATTRv患病率约为57.4/10万,中国人群的估计患病率区间为18.9/10万~74.9/10万,提示既往患病率被严重低估。随着疾病认识的深入和诊断技术的进步,ATTRv的实际疾病负担可能远超既往预期。
近年来,ATTRv治疗领域取得了突破性进展。TTR蛋白四聚体稳定剂(如氯苯唑酸、二氟尼柳、acaramidis)和基因沉默治疗药物(如依普隆特生钠、patisiran、vutrisiran、inotersen)的相继问世,使ATTRv从不可治变为可治。然而,由于该病临床表现复杂多样、异质性高,且涉及多个器官系统,单一专科难以独立完成患者的全程管理。近期发表的《遗传型转甲状腺素蛋白淀粉样变性多学科管理专家共识》(以下简称“多学科管理共识”)和《转甲状腺素蛋白淀粉样变性多学科协作筛查与诊断路径专家共识》(以下简称“筛查诊断共识”),从多学科协作视角系统阐述了ATTRv的筛查、诊断、治疗和随访规范,为推进我国ATTRv的规范化管理提供了重要指导。


我国ATTRv的临床与遗传学特征
我国ATTRv患者的临床及遗传学特征与国外存在显著差异。截至目前,我国人群中已发现40余种不同的TTR基因致病变异类型。报道较多的致病变异包括p.Val50Met、p.Ala117Ser、p.Glu74Lys和p.Gly103Arg,其中p.Val50Met在我国北方地区相对高发,p.Ala117Ser多见于南方地区。一项纳入202例中国ATTRv患者的研究显示,最常见的突变类型为Val30Met(19.8%),其次为Ala97Ser(15.8%)。
在亚型特征方面,p.Val50Met、p.Ala117Ser变异多表现为晚发型(≥50岁发病),而p.Glu74Lys、p.Gly103Arg变异以早发型(<50岁发病)为主。p.Ala117Ser患者早期腕管综合征发生率较高,p.Gly103Arg患者易出现早期眼部受累。此外,p.His108Arg变异以心肌病为主要表现,而p.Asp38Gly、p.Tyr134Cys变异患者早期可出现软脑膜受累。
我国ATTRv患者的平均起病年龄为(50.6±12.4)岁,男性患者多于女性。该病临床可累及感觉运动神经、自主神经、消化道、心脏、眼部、肾脏、软脑膜等多个系统与器官,同时累及周围神经及心脏的混合型ATTRv患者占比为30.16%~90.90%。这一高比例的混合型患者特征,凸显了多学科协作管理的重要性。
表1 ATTRv多系统临床表现
|
受累部位 |
主要临床表现 |
|
心脏 |
心力衰竭(尤以射血分数保留型多见)、主动脉瓣狭窄、心律失常(心房颤动、传导阻滞)、心源性栓塞、晕厥、心脏性猝死 |
|
周围神经 |
感觉运动神经病:肢体麻木或疼痛、分离性感觉障碍、肌肉无力、行走困难;自主神经病:体位性低血压(可致晕厥)、性功能障碍、出汗障碍、排尿障碍 |
|
消化系统 |
腹泻、便秘或两者交替;持续恶心、呕吐;不明原因体重下降;早饱感、进食后腹胀 |
|
眼部 |
瞳孔异常(花边样瞳孔、对光反射异常)、玻璃体混浊、青光眼、结膜血管异常、视乳头异常 |
|
肾脏 |
蛋白尿(可进展至肾病综合征)、肾功能不全 |
|
中枢神经系统(软脑膜) |
脑出血、蛛网膜下腔出血、脑梗死、脑积水、痴呆、癫痫、脊髓病 |
|
骨骼肌肉系统 |
双侧腕管综合征(早期常见)、肱二头肌肌腱断裂(大力水手征)、腰椎管狭窄、肩/膝/髋部疼痛 |
|
其他 |
体位性低血压(自主神经受累所致)、不明原因体重下降(需警惕消化道受累或营养不良) |
说明:ATTRv患者常同时存在多个系统受累,单一系统损害较为少见。我国混合型(同时累及周围神经及心脏)患者占比为30.16%~90.90%,临床评估时需进行全面系统的筛查。
筛查与诊断:多学科协作的前沿阵地
高危临床情景识别
ATTR起病隐匿,临床表现异质性大且缺乏特异性,加之我国临床医务人员对该病的认识及临床经验不足,导致漏诊、误诊、诊疗延迟率高,患者预后差。为实现早期诊断,筛查诊断共识明确了七类ATTR高危临床情景,包括:(1)有ATTR家族史,或存在不明原因疾病家族史;(2)无症状的TTR基因致病突变携带者;(3)不明原因心力衰竭(尤其是射血分数保留的心力衰竭);(4)不明原因左心室肥厚;(5)年龄≥65岁且合并低流速、低压力阶差的主动脉瓣狭窄,或年龄≥65岁的严重主动脉瓣狭窄患者;(6)不明原因的慢性进行性神经病;(7)快速进展的多发性神经病,且对神经病变特定治疗方法无应答。
警示征评估体系
筛查诊断共识建立了分层警示征评估体系。以心脏表现为主者,若存在不明原因左心室壁厚度≥12 mm,且合并至少1条ATTR-CM警示征(如与心力衰竭严重程度不呈比例的BNP/NT-proBNP升高、心电图肢体导联低电压、超声心动图“心尖保留”征象等),应怀疑ATTR-CM可能。以神经表现为主者,若存在排除常见病因的慢性进行性感觉或感觉运动神经病,且合并至少1条ATTRv-PN警示征(如自主神经病、腕管综合征、不明原因体重下降等),应怀疑ATTRv-PN可能。
分层诊断路径
ATTR的诊断需依托多种检查方法的互补与联合应用,遵循“先无创、后有创”的原则。主要检查手段包括生物标志物检测(BNP/NT-proBNP、高敏心肌肌钙蛋白、血清游离轻链、血清/尿蛋白免疫固定电泳)、无创多模态影像检查(心电图、超声心动图、心脏磁共振、放射性核素骨显像、神经电生理检查)、组织活检及TTR基因检测。
ATTR-CM的诊断标准为:放射性核素骨显像显示心肌摄取量达2级或3级,同时血清游离轻链、血清/尿蛋白免疫固定电泳未检出单克隆蛋白,且超声心动图或心脏磁共振检查存在任一典型心脏淀粉样变性特征,即可确诊。ATTRv-PN的诊断则需基因检测明确存在TTR基因致病突变,同时合并无法用其他原因解释的进行性长度依赖性感觉或感觉运动多发性神经病或自主神经病。
治疗策略:从对症到对因
对症治疗
ATTRv的对症治疗需根据各系统损害进行个体化干预。对于感觉运动神经病,建议进行康复治疗和针对性护理,神经病理性疼痛可参考相关治疗指南选择度洛西汀、加巴喷丁、普瑞巴林等药物。心血管系统方面,心功能不全可给予小剂量袢利尿剂或盐皮质激素受体拮抗剂;合并心房颤动/房扑的患者均应考虑抗凝治疗;症状性体位性低血压首选非药物治疗,疗效不佳时可选用米多君、屈昔多巴等药物。消化系统症状需系统性评估患者的症状及营养状况,针对胃排空障碍、便秘、腹泻等采取相应药物治疗。
病因治疗
TTR蛋白四聚体稳定剂:氯苯唑酸是目前临床应用最广泛的TTR蛋白四聚体稳定剂。对于ATTRv-PN I期患者,推荐应用氯苯唑酸葡胺软胶囊20 mg/d;对于ATTRv-CM患者及混合型ATTRv患者,推荐应用氯苯唑酸软胶囊61 mg/d。研究显示,ATTR-CM患者口服氯苯唑酸30个月后显著降低了全因死亡率和心血管病相关住院率。二氟尼柳作为另一种TTR蛋白四聚体稳定剂,在其他病因治疗不可及时可作为超适应症用药选择。Acaramidis是一种新型TTR蛋白四聚体稳定剂,已在美国、欧盟获批用于ATTR-CM的治疗,目前尚未在国内上市。
基因沉默治疗:基因沉默类药物通过抑制信使RNA表达减少TTR蛋白生成。目前仅有依普隆特生钠在我国获批用于ATTRv-PN患者的治疗,推荐剂量为每月1次、每次45 mg皮下注射。Patisiran、vutrisiran、inotersen已在国外获批用于ATTRv-PN的治疗,其中vutrisiran近期已在美国、欧盟获批用于ATTR-CM的治疗。
肝脏移植:对于起病年龄<50岁、周围神经病分期为I期的ATTRv患者,如药物治疗效果不佳,在充分权衡利弊后可考虑肝脏移植。p.Val50Met变异患者肝脏移植效果优于非p.Val50Met变异患者。
多学科管理与长期随访
多学科管理团队的建设
鉴于ATTRv临床表现复杂多样、涉及多个器官系统,多学科管理共识建议成立ATTRv多学科管理团队,成员可包括神经内科、心血管内科、消化内科、眼科、肾脏内科、罕见病科、医学遗传科、医学影像科、核医学科、临床营养科、药剂科、康复医学科、生殖医学科等。筛查诊断共识进一步建议有条件的医院建设ATTR专病中心或专病门诊,提供全面、规范的多学科协作诊疗服务与患者全程管理。同时,应推动ATTR分级诊疗,充分发挥不同级别医院的协同作用。
随访监测规范
多学科管理共识建议ATTRv患者每6个月至1年进行1次随访,重点关注对因治疗及对症治疗的效果、不良反应以及患者的依从性,同时关注各系统受累的进展情况。随访内容应包括完整的病史和查体、BMI评估、神经功能损伤评分(NIS)、复合自主神经症状评分-31(COMPASS-31)、6分钟步行试验(6MWT),以及多系统受累相关的辅助检查。
无症状基因携带者的管理
ATTRv具有常染色体显性遗传的特点,先证者诊断后,其他家庭成员可能携带TTR致病变异。多学科管理共识建议,先证者家族中可能携带TTR致病变异的亲属接受专业的遗传咨询,充分权衡利弊后自主决定是否接受基因检测。对于无症状基因携带者,首次就诊时应进行疾病相关的系统性评估并留取基线资料,随后定期随访。确认无症状基因携带者是否已经发病,需由对ATTRv具有丰富诊断和管理经验的临床医师或多学科诊疗团队进行判断。
结语与展望
ATTRv是一种可治的罕见遗传性多系统疾病。我国ATTRv患者的临床及遗传学特征与国外存在显著差异,混合型患者比例高,这决定了不能简单照搬国外指南进行管理。近期发表的两部专家共识从多学科协作视角出发,结合我国患者的临床及遗传学特点,系统阐述了ATTRv的筛查、诊断、治疗和随访规范,为推进我国ATTRv的规范化管理提供了重要指导。ATTRv的诊疗仍面临诸多挑战。疾病认知不足导致的高漏诊率和误诊率、新药的可及性和医保覆盖问题、基层医院与专病中心之间的转诊衔接等,都是亟待解决的问题。未来,随着人工智能辅助的影像学分析技术、正电子发射断层扫描等新技术的应用,以及基因编辑疗法的不断发展,ATTRv的早期诊断和治疗水平有望进一步提升。多学科协作诊疗模式的深入推进,将为ATTRv患者带来更优质的全程管理服务,改善患者的长期预后和生活质量。
参考文献
-
Delgado D, et al. Epidemiology of transthyretin (ATTR) amyloidosis: a systematic literature review[J]. Orphanet J Rare Dis. 2025,20(1):29.
-
中华医学会罕见病分会,北京医学会罕见病分会.遗传型转甲状腺素蛋白淀粉样变性多学科管理专家共识[J].中华罕见病杂志,2026,54(2):214-222.
-
国家放射与治疗临床医学研究中心.转甲状腺素蛋白淀粉样变性多学科协作筛查与诊断路径专家共识[J].中华心血管病杂志,2026,54(2):146-156.
-
Yongsheng Z, et al. Prevalence estimation of ATTRv in China based on genetic databases[J]. Front Genet,2023,14:1126836.
-
Chu X, et al. A multicenter study of hereditary transthyretin amyloidosis in China[J]. Ann Neurol,2025,97(6):1158-1167.
-
Maurer MS, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy[J]. N Engl J Med,2018,379(11):1007-1016.
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP
