首页 > 疾病防控/ 正文
深度解析医学证据,lxfs.net为你支撑决策
畸胎癌肉瘤是一种罕见的混合性恶性肿瘤,由癌成分、肉瘤成分和未成熟神经上皮成分共同构成。虽然畸胎癌肉瘤最常发生于鼻窦区域,但其在生殖道中的发生极为罕见。据研究人员所知,目前尚无关于畸胎癌肉瘤发生于外生殖器模糊患者的已发表报道。本文描述了一位 29 岁表型为男性、伴有外生殖器模糊的患者,其因腹腔盆腔巨大肿块导致肠梗阻而就诊。术中发现包括发育不全的子宫、输卵管和卵巢,均与肿瘤一并整块切除。组织病理学检查显示腺癌伴有黏液样和鳞状分化,与肉瘤区域、未成熟神经上皮及软骨紧密混合。独特的形态学特征及PAX8阳性表达对于确认肿瘤起源于苗勒管、并将其与伴有体细胞型恶性肿瘤的畸胎瘤及其他生殖细胞肿瘤相鉴别具有关键作用。对侧卵巢表现为成纤维细胞性间质,缺乏卵泡和管状结构。这些发现支持了畸胎癌肉瘤可能起源于原始胚胎组织发育停滞的假说,这也是该病提出的组织发生机制之一。本病例中检测到具有临床干预意义的PIK3CA突变,鉴于患者对传统铂类化疗反应不佳,这提示了一个潜在的治疗靶点。
背 景
畸胎癌肉瘤是由癌成分、肉瘤成分和神经外胚层成分构成的侵袭性肿瘤。该病于 1983 年由Shanmugaratnam等人首次在鼻窦区域描述,截至 2014 年,报道病例不足 150 例。具有相似组织学特征的病例报告已见于肺、颅内、甲状腺、宫颈、子宫和卵巢,但据研究人员所知,尚无发生于发育不良卵巢的病例报道。对畸胎癌肉瘤起源的有限认识可能导致治疗效果欠佳。本病例报告展示了诊断性形态学特征,并讨论了与诊断和预后相关的潜在标志物。
病 例
一位 29 岁表型为男性、伴有外生殖器模糊的患者,因腹痛、腹胀及排气困难1.5个月就诊。生殖器检查显示小阴茎、尿道下裂、阴囊分叉,双侧睾丸未触及。肛门前方可见一开口,怀疑为阴道口。直肠指检显示直肠前壁隆起,结肠黏膜光滑。影像学检查发现膀胱与直肠之间有一巨大肿块,导致乙状结肠和小肠移位,并压迫双侧输尿管。未检测到睾丸或卵巢结构。术前血清甲胎蛋白和乳酸脱氢酶水平升高,总结于表1。以生殖细胞肿瘤的初步诊断进行了剖腹探查术。术中发现一管状结构(可能为子宫)附着于肿块前表面,并与乙状结肠一并切除。随后进行了回结肠吻合术和回肠吻合术。
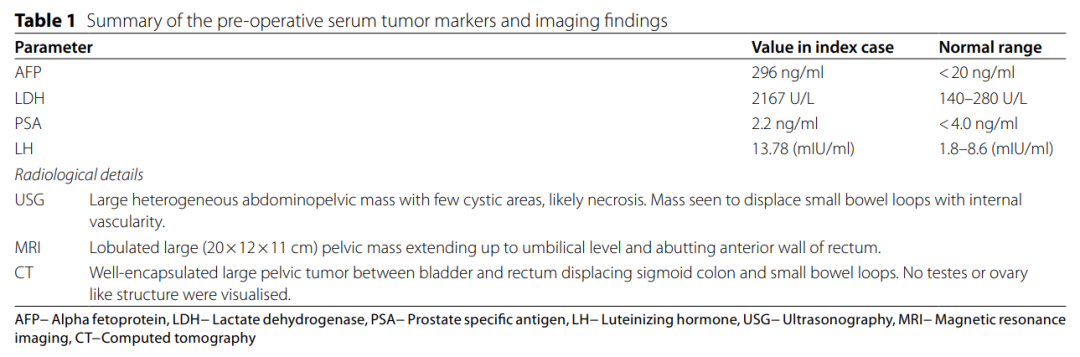
表1 术前血清肿瘤标志物和影像学检查结果汇总
大体观察显示肿块重 5 公斤,大小为 28×26×6 cm,切面呈实性囊性、灰白色,伴有出血和坏死(图1a)。子宫、输卵管及附着的卵巢被确认附着于肿块表面,但未被肿块浸润。而直肠乙状结肠则被浸润至浆膜下层。一侧可见条索状卵巢,缺乏卵泡;子宫内膜、子宫肌层及输卵管组织学上未见异常(图1b和c)。
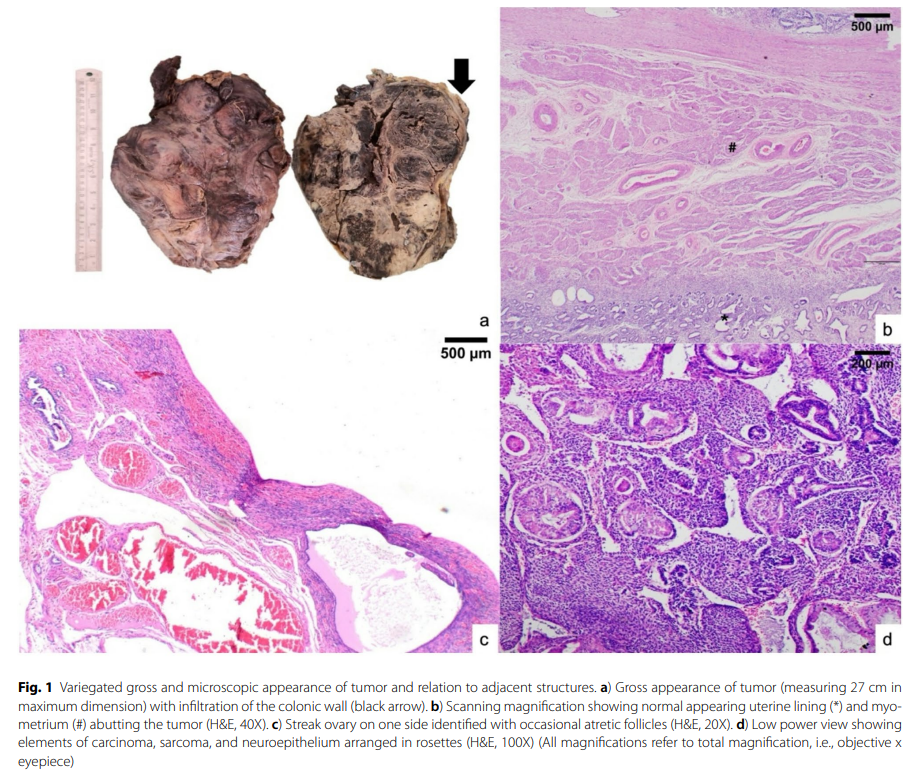
图1 肿瘤的大体和显微镜下形态多样,并与邻近结构存在关联
肿瘤在组织学上重现了其多样的大体外观,表现为恶性上皮成分、间叶成分与未成熟胎儿组织相互混杂(图1d)。上皮成分为苗勒管腺癌,以PAX8阳性为证据,伴有黏液样分化和桑葚样化生。可见显著的核上及核下空泡,类似于胎儿型内胚层分化,并表达干细胞标志物Sal样蛋白4。这些细胞对激素受体呈阴性,PTEN和错配修复蛋白表达保留,p53免疫染色呈野生型。肉瘤区域大部分为未分化成分,但可见散在的未成熟软骨岛,缺乏其他异源性成分,如横纹肌母细胞、脂肪细胞和成骨细胞(图2)。
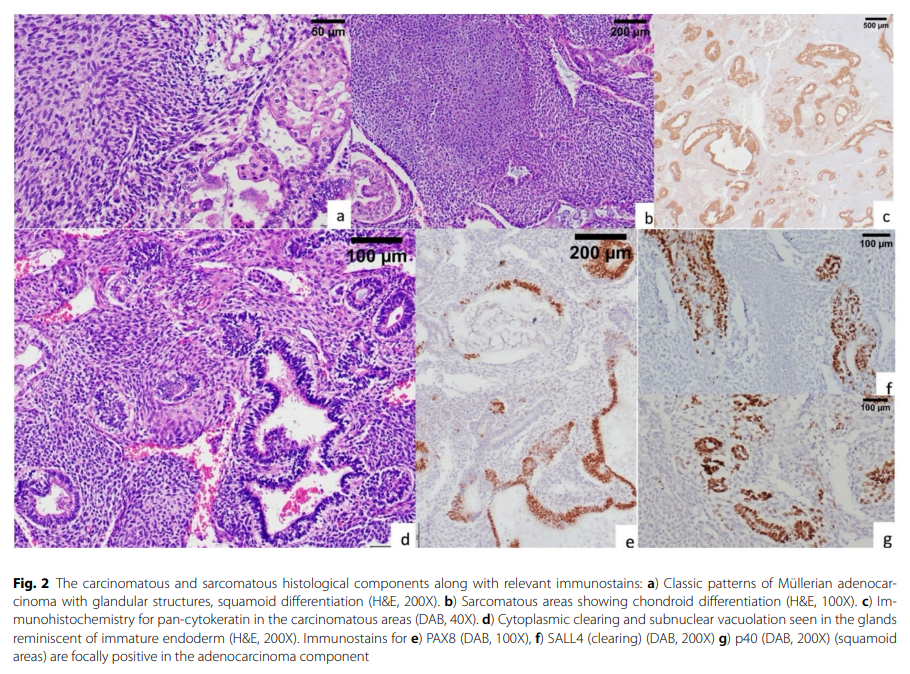
图2 癌性和肉瘤性组织学成分及其相关免疫组化染色结果
第三种细胞类型是呈玫瑰花环状和片状排列的小圆细胞,这些细胞突触素、CD56和SALL4染色阳性,代表未成熟神经上皮或"原始神经外胚层",这对于区分畸胎癌肉瘤与恶性苗勒管混合瘤至关重要(图3)。尽管进行了充分取材,但未见成熟生殖细胞衍生成分,且glypican-3、CD30、CD117和SOX2均为阴性,这些结果不支持生殖细胞肿瘤的诊断。INI1和BRG1免疫染色显示所有细胞类型均保留核表达,而β-catenin呈通常的膜性染色。
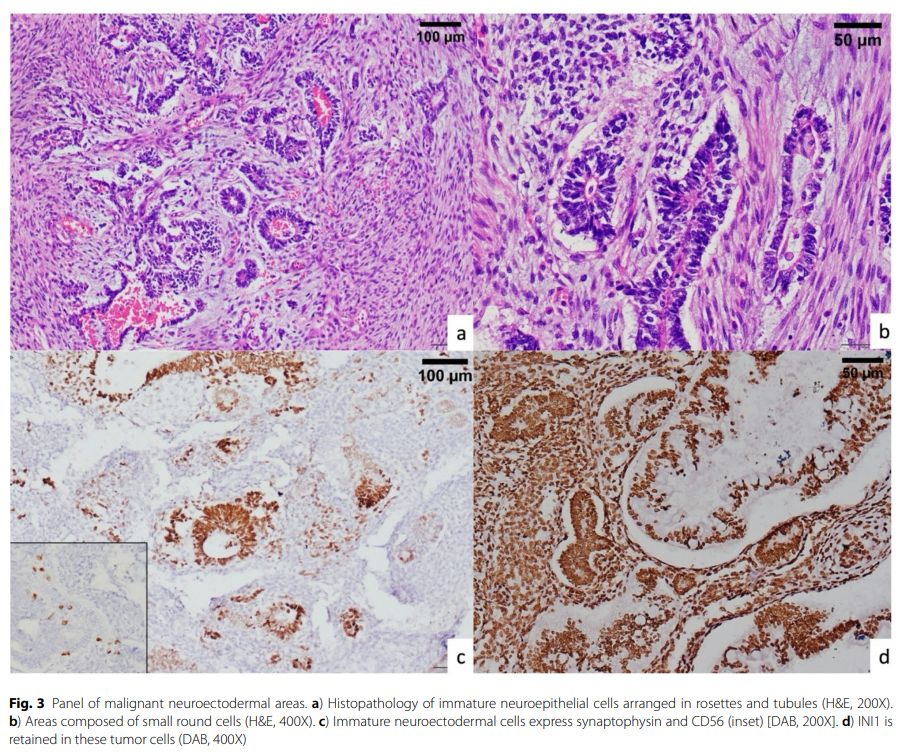
图3 恶性神经外胚层区域图谱
研究人员使用试剂盒进行PIK3CA突变检测。引物和探针组设计用于检测涉及PIK3CA的突变:E542K、E545A、H1047Y或C420R,并设有内部对照。研究人员发现了一个p.Q546R错义突变,导致第9号外显子第1637位核苷酸由A变为G。使用针对18号、X和Y染色体的着丝粒探针,对福尔马林固定石蜡包埋肿瘤组织进行荧光原位杂交,结果显示存在两个不同的细胞群体。计数至少 100 个非重叠细胞核,其中约 80% 的细胞核显示单个X信号、两个18号染色体信号且无Y信号,符合XO细胞特征;而 20% 显示一个X和一个Y信号以及两个18号染色体信号,符合XY细胞特征,提示45XO/46XY嵌合体(图4)。术后病程平稳,在获得明确组织病理学诊断之前,鉴于临床和影像学初步印象为生殖细胞肿瘤,患者接受了博来霉素、依托泊苷和顺铂辅助化疗。4 个周期后随访显示甲胎蛋白水平轻度升高(19.4 ng/ml),但无临床或影像学疾病进展证据。
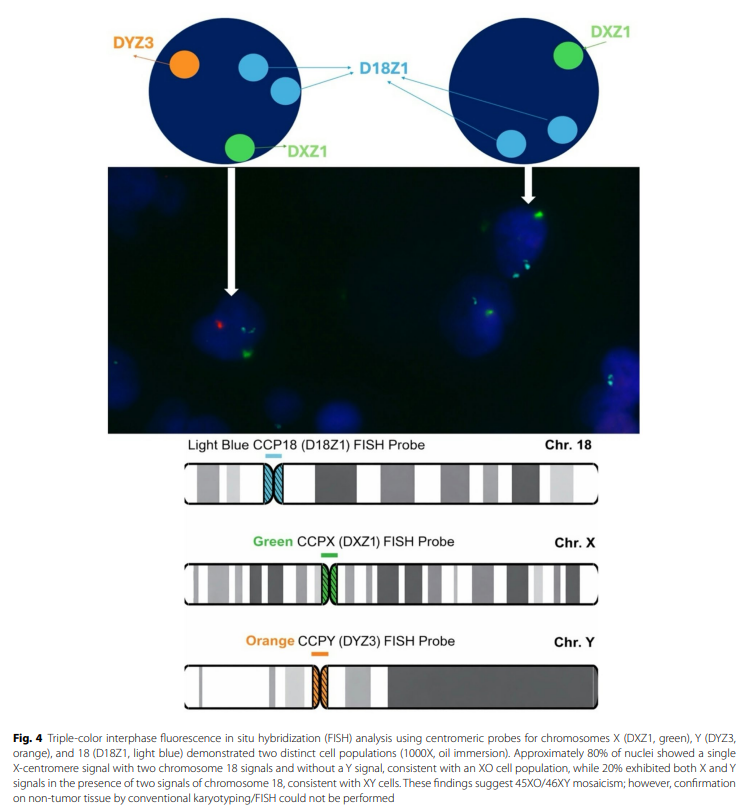
图4 使用针对X染色体(DXZ1,绿色)、Y染色体(DYZ3,橙色)和18号染色体(D18Z1,浅蓝色)着丝粒探针的三色间期荧光原位杂交(FISH)分析显示存在两种不同的细胞群(1000倍,油镜)
讨 论
畸胎癌肉瘤是性腺神经系统(SNT)的罕见肿瘤,以多表型分化和有争议的组织发生为特征。目前已提出多种起源假说,包括:①原始胚胎组织发育停滞,②癌的去分化,以及③上皮-间充质转化。据研究人员所知,这是首例发生于性腺发育不全的畸胎癌肉瘤病例报告,支持其通过异常胚胎发生起源的观点。
首例卵巢畸胎癌肉瘤由Ehrmann等人于 1990 年报道。对具有相似形态的卵巢肿瘤进行文献回顾,共发现 9 例,总结于表2。这些肿瘤曾使用多种术语,如"畸胎样癌肉瘤"、"伴有恶性神经外胚层成分/分化的恶性苗勒管混合瘤"、"伴有卵黄囊瘤的卵巢癌肉瘤"以及"伴有腺肉瘤的子宫原始神经外胚层肿瘤"。
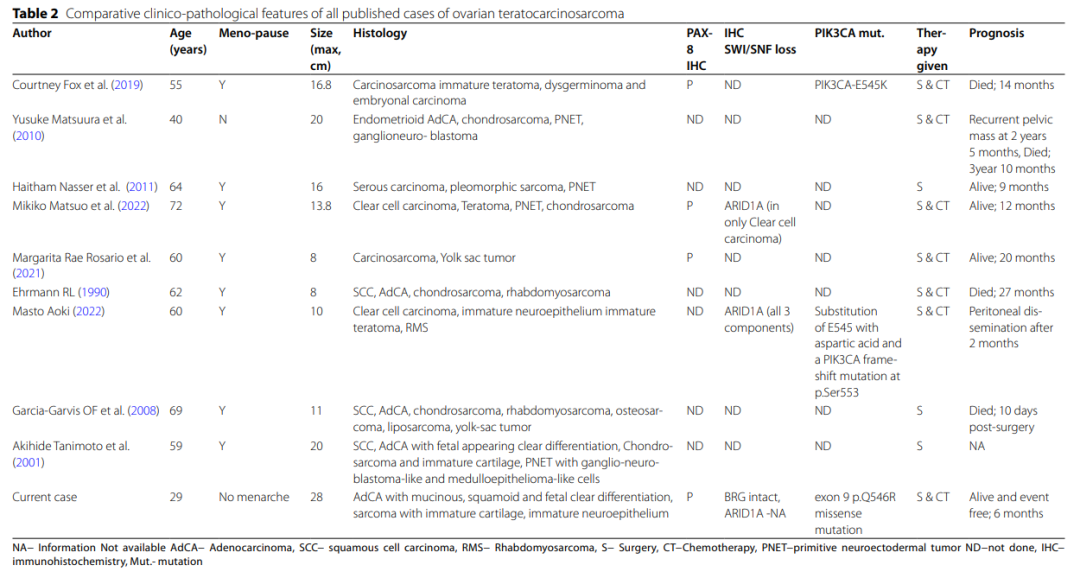
表2 已发表的卵巢畸胎癌肉瘤病例的临床病理特征比较
未成熟神经上皮、苗勒管癌和肉瘤成分的混合,支持卵巢畸胎癌肉瘤的诊断,并有助于将其与恶性苗勒管混合瘤、未成熟畸胎瘤及伴有体细胞型恶性肿瘤的畸胎瘤相鉴别。虽然癌向未成熟神经上皮的化生性转化已在癌肉瘤/恶性苗勒管混合瘤中有所描述,但未成熟软骨与未成熟神经上皮同时存在,支持真正的生殖细胞起源而非化生。此外,广泛取材未见良性器官样结构,这排除了本病例中存在共存成熟畸胎瘤的可能。
对于腹腔广泛受累的病例(如本例),需要借助PAX8等免疫染色来确认苗勒管起源。未成熟标志物SALL4可在伴有胞质透亮的癌区域表达,应谨慎解读以避免误诊为卵黄囊瘤。BRG1和β-catenin的突变免疫表型被列为鼻窦畸胎癌肉瘤的特征性分子改变;然而,四分之一病例显示表达保留。这些标志物的缺失需要在卵巢畸胎癌肉瘤中进行评估,迄今为止,已有两例卵巢畸胎癌肉瘤病例显示SWI/SNF复合体另一成员ARID1A的缺失。文献检索发现,在鼻窦畸胎癌肉瘤和两例卵巢畸胎癌肉瘤病例中(在进行了检测的病例中)均存在PIK3CA致病性突变,随后对福尔马林固定石蜡包埋组织进行了同样的检测。据作者所知,p.Q546R PIK3CA突变在任何部位的畸胎癌肉瘤中均未见报道。
手术、化疗以及少数情况下的免疫治疗联合方案已尝试使用,但效果有限。从表2可以看出,大多数卵巢畸胎癌肉瘤病例确诊时已处于晚期,患者于诊断后 3 年内死亡,反映了其不良的临床预后。PIK3CA可能是一个潜在的可预测生物标志物,而哺乳动物雷帕霉素靶蛋白抑制剂的作用仍有待探索。对肿瘤组织进行的荧光原位杂交提示45XO/46XY嵌合体。然而,由于无法通过常规核型分析及对非肿瘤组织进行荧光原位杂交分析来排除组织限制性嵌合体并确定体质性染色体状态,这是本研究的一个局限性。此外,由于该抗体不可获取,未能进行ARID1A免疫组织化学检测。
畸胎癌肉瘤发生于发育不良性腺,支持了这些肿瘤起源于原始胚胎组织发育停滞的假说。诊断虽具挑战性,但可通过以下特征确立:杂乱排列的未成熟神经上皮,以及伴有胞质透亮、表达PAX8和SALL4的癌成分。如本病例中观察到的具有临床干预意义的PIK3CA突变,可成为对含铂化疗方案反应不佳病例的潜在治疗靶点。
参考文献:
Sood, R., NS, B.P., Dash, N.R. et al. Teratocarcinosarcoma arising in dysgenic gonads: an unusual location for a rare tumor. Surg Exp Pathol 9, 22 (2026). https://doi.org/10.1186/s42047-026-00237-4
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP
