深度解析医学证据,DeepEvidence为你支撑决策
嵌合抗原受体T细胞(CAR-T)治疗已经彻底改变了复发/难治性B细胞急性淋巴细胞白血病(R/R B-ALL)等血液恶性肿瘤的治疗格局,目前全球已有多种CAR-T产品获批用于R/R B-ALL。尽管初始缓解率很高,但仍存在重大挑战,包括因抗原逃逸和CAR-T细胞持久性有限导致的疾病复发;治疗相关不良事件如细胞因子释放综合征、免疫效应细胞相关神经毒性综合征、血液学毒性和感染;以及CAR-T治疗的可及性有限。
《Biomarker Research》近日发表综述,中国医学科学院血液血液病医院(中国医学科学院血液学研究所)王迎教授和查晨宇教授讨论了CAR-T的局限性及克服这些局限性的策略,特别是在B-ALL的背景下,包括异基因CAR-T、双靶点CAR-T以及与新技术和药物的联合策略。此外还探讨了CAR-T治疗后巩固性异基因造血干细胞移植的作用,以及将CAR-T整合到B-ALL一线治疗中的潜力。作者认为,未来研究应致力于提高CAR-T的疗效、降低毒性、改善可及性,并将其应用扩展到B-ALL的更早线治疗。

背景
嵌合抗原受体T细胞(CAR-T)治疗的出现显著改善了复发/难治性(R/R)B-ALL患者的预后。截至2026年4月,美国已有三种CAR-T产品获批用于治疗R/R B-ALL:Tisagenlecleucel(tisa-cel,2017年)、Brexucabtagene Autoleucel(brexu-cel,2021年)和Obecabtagene Autoleucel(obe-cel,2024年)。三种CAR-T产品实现了71%至81%的完全缓解(CR)率,显示出良好的长期反应。
在中国,由中国医学科学院血液病医院(中国医学科学院血液学研究)开发的Inaticabtagene Autoleucel(纳基奥仑赛,inati-cel,2023年)成为中国首个获批用于成人R/R B-ALL的CAR-T产品。在关键临床试验中,Inati-cel实现了85.4%的CR率,2年无复发生存(RFS)率为35.8%,2年总生存(OS)率为55.2%。重庆精准生物的普基仑赛2025年11月获批上市,治疗儿童及青少年R/R B-ALL。此外,越来越多的CAR-T治疗R/R B-ALL的临床试验正在进行中。
尽管这些高初始缓解率令人鼓舞,但仍存在重大挑战,包括因抗原逃逸和CAR-T持久性有限导致的治疗失败、治疗相关毒性以及可及性有限。作者讨论了这些挑战以及在R/R B-ALL背景下旨在克服它们的最新研究进展。
R/R B-ALL中CAR-T细胞治疗的挑战
抗原丢失
抗原丢失主要源于肿瘤细胞内部的机制(图1),这可导致CD19阴性复发,是R/R B-ALL患者接受CD19 CAR-T治疗后治疗失败和长期生存差的主要原因。
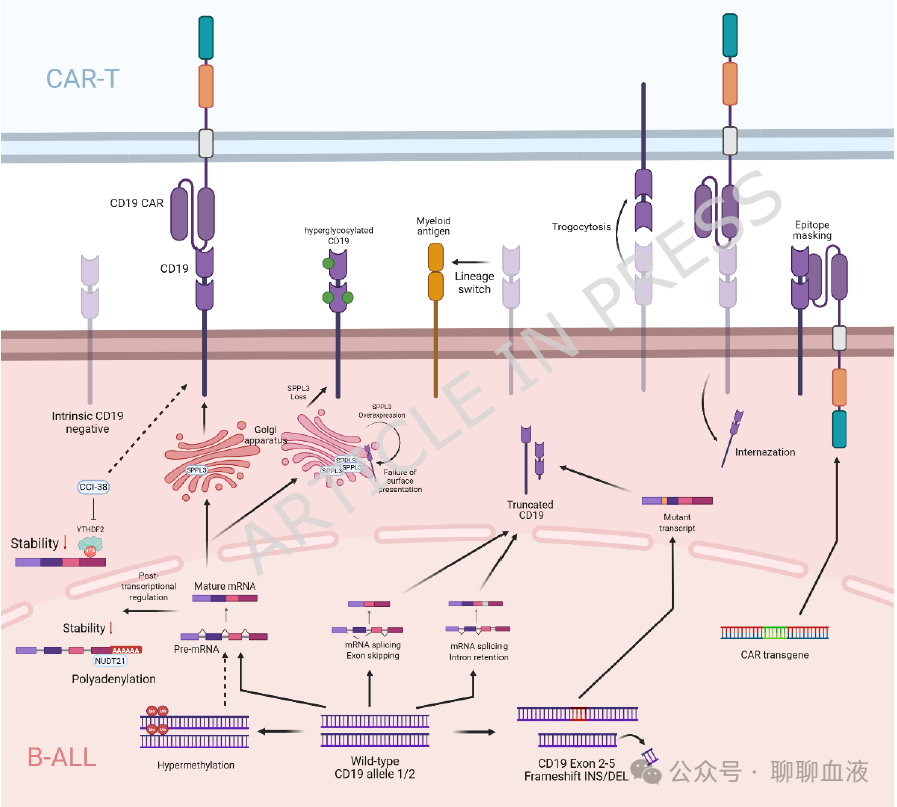
图1.B-ALL中 CD19逃逸机制概述
B-ALL细胞可通过多种机制下调或丢失CD19抗原表达:
在DNA水平,CD19外显子2-5的获得性移码突变可导致截短的CD19蛋白缺乏膜锚定,使其在细胞表面无法被CD19 CAR-T细胞和流式细胞术检测到。CD19基因启动子的超甲基化也可降低CD19表达,阻止CD19 CAR-T细胞的识别。
在RNA水平,B-ALL中CD19 mRNA的可变剪接产生无法编码功能性CD19蛋白的异构体,损害CD19 CAR-T识别并促进复发。RNA结合蛋白YTHDF2可通过m6A依赖性mRNA降解下调CD19表达,导致CD19 CAR-T治疗的抗原逃逸。
在翻译后水平,CD19的糖基化失调也可能影响B-ALL细胞对CD19 CAR-T的识别。
淋巴-髓系谱系转换,这在KMT2A重排ALL中尤为常见,是CAR-T免疫压力下由谱系可塑性驱动的另一种抗原阴性复发机制。
在CAR-T治疗前已在部分ALL患者中鉴定出CD19阴性白血病原始细胞的预先存在亚克隆。在CAR-T细胞的选择性压力下,这些阴性亚克隆可能扩增并最终复发。
CAR-T制造过程中CD19 CAR转基因意外转导到B-ALL原始细胞中,随后回输到患者体内,也可导致CD19阴性复发。这是由于CAR在原始细胞表面表达后发生顺式结合并掩蔽CD19表位,阻止CAR-T细胞和流式细胞术的识别。
CAR-T细胞识别和激活依赖于靶抗原的高密度表达。因此,B-ALL原始细胞上抗原的下调可能使免疫逃逸成为可能。既往CD19靶向治疗(如贝林妥欧单抗)可导致CD19下调。此外,研究表明B-ALL细胞与CD19 CAR-T细胞的相互作用诱导CAR介导的CD19内化和降解。此外,CD19 CAR-T细胞的胞啃作用可将CD19从B-ALL细胞转移到自身,降低B-ALL细胞上的CD19密度并损害CAR-T介导的杀伤。有趣的是,CD19单克隆抗体可促进CD19 CAR-T细胞从B-ALL细胞的快速解离,减轻胞啃作用诱导的CD19丢失。CD19 CAR-T与抗CD19 mAb的联合具有协同抗B-ALL效应,但支持这种联合的临床证据有限。
CAR-T功能与持久性有限
CAR-T的内在因素(如功能受损、扩增不足、持久性差和耗竭)也对治疗疗效构成重大障碍,可导致难治性疾病或抗原阳性复发。从制造到输注的每个步骤都可能影响体内活性,而肿瘤微环境可进一步加剧这些问题。
T细胞分化阶段对CAR-T细胞持久性有显著影响。来源于早期分化阶段(初始T细胞、中央记忆T细胞和干细胞样记忆T细胞)的CAR-T细胞表现出优越的扩增和持久性,并与良好预后相关。相反,终末分化T细胞与扩增差和复发相关。富集早期分化T细胞的策略包括亚群选择、细胞因子添加(IL-7、IL-15和IL-21)、CAR结构修饰以及药理学或遗传学调控。在Aldoss等的研究中,采用初始/记忆T细胞选择的CAR-T实现了87%的CR/完全缓解伴不完全血液学恢复(CRi)率,1年RFS和OS率分别为52.6%和62.3%。需要进一步的临床研究来确定这些早期分化T细胞富集策略是否能为R/R B-ALL患者提供额外获益。
CAR设计也显著影响体内活性。适度降低单链可变片段(scFv)亲和力可在不损害抗肿瘤功能的情况下减轻不良效应。此外,低亲和力scFv的特征,如连续激活和与靶抗原的快速解离,可能增强CAR-T增殖信号和体内持久性。与tisa-cel和brexu-cel相比,obe-cel中的scFv具有更低的亲和力(均来源于FMC63),这可能部分解释了obe-cel毒性更低和持久性改善的原因。
另一个因素为scFv的免疫原性。鼠源scFv可诱导宿主抗CAR免疫反应,限制其功能和持久性。使用人源化scFv是解决这一问题的潜在策略,但人源化CAR-T产品是否优于鼠源产品用于治疗R/R B-ALL仍不确定。
CD28和4-1BB是最常用的共刺激分子。CD28诱导快速激活和早期活性,而4-1BB在临床前研究中显示出更优越的持久性。在一项头对头比较中,4-1BB组所有患者均达到微小残留病(MRD)阴性缓解,而CD28组为89%,4-1BB组的1年RFS和OS更高。然而相关报告存在矛盾:部分研究报告两者之间长期生存无显著差异,但也有一项真实世界研究报告tisa-cel(4-1BB)和brexu-cel(CD28)的结局相当。鉴于多种因素可影响CAR-T疗效,需要更大规模的随机对照试验来分析共刺激域选择对R/R B-ALL CAR-T治疗的影响。
输注后,CAR-T细胞依赖肿瘤抗原刺激来激活其抗肿瘤效应功能,但持续的抗原暴露可驱动CAR-T耗竭,限制其活性和持久性,导致复发。大量研究证实,输注后的CAR-T细胞表现出耗竭相关的转录特征和表型,包括耗竭相关转录因子的激活和免疫检查点的上调。此外,抗原暴露启动CAR-T细胞的表观遗传重编程。通过对R/R B-ALL患者CD8+ CAR-T细胞甲基化景观的纵向分析,Zebley等报告了CD8+ CAR-T细胞中的效应基因在输注后第一周内获得去甲基化,而记忆相关基因逐渐发生甲基化沉默。同时,CAR-T细胞向耗竭表型转变,如CX3CR1、BATF和TOX等位点的去甲基化所证明,最终限制其持久性。
血液恶性肿瘤的免疫抑制微环境可通过多种机制削弱CAR-T抗肿瘤活性和持久性,是CAR-T耐药或复发的重要因素。多项研究报告R/R B-ALL患者中高基线水平的免疫抑制细胞,如调节性T细胞(Tregs)、髓源性抑制细胞和抑制性中性粒细胞,与不良预后相关,提示这些抑制细胞是潜在的治疗靶点。
此外,检查点配体-受体相互作用导致共抑制信号的传递,靶向免疫检查点可能改善CAR-T疗效,尽管在B-ALL中的证据有限。
不良事件
细胞因子释放综合征(CRS)
托珠单抗和皮质类固醇是CRS管理的基石。托珠单抗可有效控制CRS,而不损害B-ALL患者体内CAR-T扩增、缓解率或长期结局。在轻度CRS症状出现时早期使用托珠单抗,或在高危人群中预防性使用,可能减轻严重CRS风险。
皮质类固醇用于托珠单抗后未改善的CRS,但存在可能损害CAR-T疗效的担忧。然而,多项研究表明,皮质类固醇的使用(即使早期启动)不会影响B-ALL患者的CAR-T扩增或疗效。考虑到类固醇的额外负面影响(如增加感染风险),建议将皮质类固醇治疗的剂量和持续时间降至最低有效水平,同时确保疗效。对于难治性CRS,可考虑IL-6抑制剂司妥昔单抗、IL-1受体拮抗剂anakinra和JAK抑制剂芦可替尼等选择,但相关证据有限。
研究努力也集中在从源头减轻CRS和其他毒性,包括优化CAR-T输注方案和设计毒性更低的CAR-T构建体。在FELIX研究中,研究者根据骨髓疾病负荷调整给药方案,并在第1天和第10天分次给予obe-cel,最终仅报告3例(2.4%)≥3级CRS和9例(7.1%)≥3级免疫效应细胞相关神经毒性综合征(ICANS)。在2期CART19-BE-02试验中,varnimcabtagene autoleucel(var-cel)以四个递增剂量(0.1、0.3、0.6和2×106 CAR-T细胞/kg)分次给药。在32例成人R/R B-ALL中,≥3级CRS的发生率仅为12%,任何级别ICANS为3%。这些发现突出了个性化剂量调整和分次给药增加CAR-T安全性的潜力。
低毒性CAR-T细胞的设计涉及CAR结构的修饰和CAR-T细胞的基因工程。Obe-cel利用低亲和力scFv,有助于降低毒性。如前所述,obe-cel在FELIX试验中显示出良好的安全性,工程化CAR-T细胞过表达膜结合受体或分泌细胞因子阻断剂等策略可帮助中和循环促炎细胞因子。此外,ssCART-19细胞(一种携带IL-6靶向短发夹RNA的抗CD19 CAR-T)的1期试验报告≥3级CRS为17.6%,无ICANS,3个月客观缓解率(ORR)为64.7%,提示IL-6敲低可能在不损害疗效的情况下增强安全性。未来需要头对头试验验证ssCART-19细胞在安全性方面优于传统CAR-T。
神经毒性(ICANS)
ICANS的危险因素与CRS重叠,包括高疾病负荷、高CAR-T剂量、高峰值扩增、强化淋巴细胞清除、升高的促炎细胞因子和内皮损伤标志物。CRS的严重程度也与ICANS相关。独特的预测因素包括既往神经系统合并症,以及反映轴突损伤和ICANS严重程度的生物标志物如神经丝轻链(NfL)和胶质纤维酸性蛋白。
ICANS的标准治疗为皮质类固醇治疗,而对于皮质类固醇难治性ICANS尚无标准化管理。鉴于CRS和ICANS共享的病理生理学,一些用于难治性CRS管理的策略可改编用于难治性ICANS。IL-1受体拮抗剂anakinra常被视为难治性ICANS的一线选择,因为多项回顾性研究显示其有效且不损害CAR-T疗效。在Park等的前瞻性研究中,预防性anakinra给药也降低了淋巴瘤患者ICANS的发生率。司妥昔单抗在最近的回顾性研究中也显示出疗效,但前瞻性研究的支持有限。
血液学毒性
免疫效应细胞相关血液学毒性(ICAHT)指CAR-T细胞治疗后发生的血细胞减少。ICAHT的发病机制是多因素的,源于基线造血储备、既往治疗、淋巴细胞清除和CAR-T介导的骨髓炎症。
多种危险因素与ICAHT相关,详见其他综述。此外,Nair等最近开发了ALL-Hematotox(ALL-HT)评分系统来预测R/R B-ALL患者CAR-T后的血液学毒性,该系统纳入基线骨髓疾病负荷、CRP水平、绝对中性粒细胞计数、血小板计数和血红蛋白水平,可有效预测了B-ALL患者严重中性粒细胞减少的发生[曲线下面积(AUC)=0.84]。Liang等也开发了两个早期严重ICAHT的预测模型:eIPM Pre(基于输注前因素)和eIPM Post(用CAR-T后第3天铁蛋白替代输注前铁蛋白)。它们在R/R B-ALL患者中的AUC分别为0.87和0.88,显示出良好的区分能力。识别危险因素并应用预测模型可促进ICAHT患者的早期识别,实现后续管理并降低非复发死亡率。
ICAHT的治疗仍处于探索阶段,基于现有经验,输血治疗和造血生长因子是潜在选择。粒细胞集落刺激因子(G-CSF)通常用于缩短CAR-T治疗后中性粒细胞减少的持续时间。淋巴瘤患者中早期/预防性G-CSF已被证明可安全有效地降低中性粒细胞减少的发生率或加速中性粒细胞恢复,但在B-ALL患者中未观察到这些获益。鉴于这些证据基于回顾性研究,需要前瞻性临床试验来研究G-CSF的安全性和最佳给药策略。对于难治性ICAHT,低剂量皮质类固醇和高剂量静脉免疫球蛋白(IVIG)可能有效,但证据有限。如果有供者可用,输注供者造血干细胞以恢复造血是另一种选择,但最佳细胞剂量和时机需要进一步确定。
低丙种球蛋白血症
在B-ALL患者中,低丙种球蛋白血症(hypogammaglobulinemia,HG)是CD19 CAR-T治疗脱靶毒性的表现。HG意味着体液免疫缺陷,显著增加感染风险。当前管理依赖于血清IgG监测和免疫球蛋白替代,更积极的策略通常保留用于儿童患者。
IEC-HS
部分接受CAR-T治疗的患者发生噬血细胞性淋巴组织细胞增多症(HLH)样疾病,称为免疫效应细胞相关噬血细胞性淋巴组织细胞增多症(IEC-HS),可导致严重的多器官功能障碍。尽管IEC-HS常在CRS消退期间或之后发生,并与CRS共享重叠的发病机制和临床特征,但被视为需要不同治疗方法的独立实体。需要仔细鉴别严重CRS和IEC-HS,血清细胞因子谱和铁蛋白水平可能有助于鉴别。
IEC-HS的治疗也缺乏前瞻性研究数据。目前推荐的一线治疗是anakinra±皮质类固醇,在2级IEC-HS发展时启动。如无改善,anakinra和皮质类固醇以增加的剂量联合使用,可考虑芦可替尼作为二线治疗。对于二线治疗难治的IEC-HS,可使用emapalumab,病例报告报道了其在控制难治性IEC-HS中的疗效。
可及性问题
尽管多种CAR-T产品获批用于R/R B-ALL,但这种变革性治疗的潜力仍受到一系列可及性障碍的限制,阻止许多符合条件的患者接受治疗。这些障碍跨越患者特异性、后勤和财务领域。理解这些挑战对于制定扩大CAR-T治疗覆盖范围的策略至关重要。
目前所有获批的CAR-T产品均来源于自体T细胞。然而考虑到患者特异性因素,自体CAR-T治疗存在若干局限性。首先,部分患者可能因严重淋巴细胞减少或临床不稳定而无法成功进行单采,这些都可能阻碍收集足够的自体T细胞。其次,即使单采可行,采集的T细胞质量可能因既往治疗和肿瘤微环境而受损。这些局限性限制了自体CAR-T在某些患者中的适用性,凸显了异基因替代方案的迫切需求。
自体CAR-T的常规制造过程从单采到产品发布需要2-4周,不包括运输或质控的额外时间。对于快速进展的R/R B-ALL患者,这一等待期与疾病进展或输注前死亡的重大风险相关,可能使其不符合输注条件。此外, 较长的静脉到静脉时间(从单采到输注)与显著较低的CR率和降低的OS相关。这些发现凸显了加快CAR-T制造和更高效后勤的迫切需求,这需要创新的制造策略,如快速即时(POC)生产或使用现成的异基因产品来缩短静脉到静脉时间。
最后,CAR-T治疗的高成本代表了可及性的另一个重大障碍。由于个性化和复杂的制造过程,获批的商业CAR-T产品的成本超过每次输注40万美元,这给患者和医疗系统带来沉重的经济负担。这种高成本也加剧了全球获取差异,因为这些产品在低收入和中等收入国家基本无法获得,即使在高收入环境中也常仅限于少数符合条件的患者。因此,降低CAR-T治疗成本是迫切需求,可通过优化制造过程来实现,如POC生产。值得注意的是,西班牙学术开发的var-cel已证明医院内制造可在保持疗效的同时降低成本,新兴的体内CAR-T生成策略有望进一步简化生产和降低成本。
髓外病变
B-ALL固有的迁移生物学赋予其浸润髓外部位的能力,常涉及中枢神经系统和睾丸等受保护的庇护所,导致髓外病变(EMD)。早期研究如ELIANA和ZUMA-3试验排除了CNS-3状态(定义为脑脊液中≥5个白细胞/μL且可识别淋巴原始细胞)的患者。然而随着研究进展,后续研究越来越多地证明CAR-T细胞可有效迁移到CNS并消除白血病原始细胞,在伴有CNS受累的R/R B-ALL患者中实现71%-95%的CR,与髓内疾病患者相当。
关于CAR-T治疗非CNS EMD的数据相对有限。既往病例报告描述了CAR-T在B-ALL患者皮肤和睾丸浸润治疗中的应用,表明CAR-T细胞对非CNS EMD也具有迁移和抗肿瘤活性。然而,非CNS EMD对CAR-T的反应模式与髓内疾病不同。Holland等观察到髓内疾病患者和非CNS EMD患者对CAR-T反应的时间和深度差异:在18例伴有非CNS EMD的R/R B-ALL患者中,3例(16.7%)需要3-6个月才能在髓外部位达到最佳反应,这显著晚于骨髓疾病通常在第28天看到的最佳反应。在14例达到骨髓CR的患者中,7例未能在其非CNS EMD病灶中达到CR。类似地,Epperly等也报告了CAR-T治疗后非CNS EMD的延迟反应或缓解深度不佳,这些差异可能归因于CAR-T细胞向EMD部位的迁移差异以及EMD微环境的影响。这些发现提示,CAR-T向EMD部位的浸润不足或EMD比骨髓更具免疫抑制性微环境。未来研究应聚焦于阐明CAR-T在EMD中的活性和动力学,并制定增强对EMD(特别是非CNS EMD)疗效的策略。
B-ALL中CAR-T治疗的新进展
通过CRISPR技术优化CAR-T
在R/R B-ALL CAR-T治疗中,使用成簇规律间隔短回文重复序列(CRISPR)/CRISPR相关蛋白9(Cas9)敲除CAR-T细胞中的特定基因可在多个维度增强治疗结局,包括疗效、安全性和可及性(图2)。
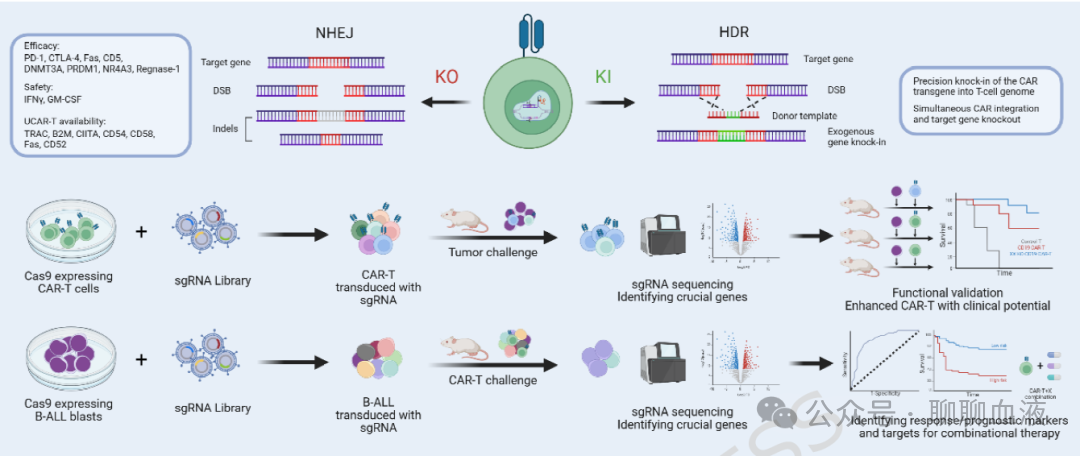
图2.在B-ALL中的,通过CRISPR敲除、敲入、敲除筛选优化CAR-T
提高疗效的核心策略涉及靶向敲除CAR-T细胞表面的抑制性分子如免疫检查点。例如,Agarwal等生成了细胞毒性T淋巴细胞相关蛋白4(CTLA-4)敲除CAR-T细胞,在B-ALL小鼠模型中显著降低肿瘤负荷并延长生存。此外,与单独免疫检查点阻断或敲除相比,CRISPR/Cas9介导的CAR-T细胞中T细胞耗竭关键调控因子的敲除可能更有效地重塑T细胞命运,赋予更持久的抗耗竭能力。这些修饰的CAR-T细胞在持续抗原暴露下维持早期表型和增殖能力,在包括B-ALL在内的多种肿瘤小鼠模型中显示出延长生存和增强抗肿瘤疗效,并展现出临床应用潜力。
安全性方面,主要关注点为CRS和ICANS。主要策略涉及使用CRISPR/Cas9敲除CAR-T细胞中参与CRS/ICANS的关键细胞因子,同时避免损害CAR-T疗效。
通过CRISPR改善CAR-T可及性的最佳例证是使用来源于健康供者的通用CAR-T(UCAR-T)细胞。CRISPR编辑使这些异基因T细胞在患者中更安全、更持久地发挥作用。为防止宿主免疫排斥,UCAR-T细胞通常通过β2-微球蛋白敲除工程化破坏人类白细胞抗原(HLA)I类表达。此外,CD52敲除使UCAR-T细胞抵抗alemtuzumab,后者用于淋巴细胞清除以清除宿主淋巴细胞,同时保留输注的UCAR-T细胞,从而增加其持久性。
除基因敲除外,CRISPR/Cas9还可通过同源定向修复实现CAR组分的靶向敲入(KI)。经典例子是将CD19 CAR整合到TRAC基因座以生成TRAC-CAR-T细胞,这种位点特异性整合确保均匀的CAR表达,内源性TRAC启动子的调控维持最佳CAR水平,减少耗竭。与随机病毒整合的传统CAR-T细胞相比,TRAC-CAR-T细胞在B-ALL小鼠模型中实现了更持久的疾病控制。另一种策略是将CD19 CAR整合到PD1基因座(PD1-CAR-T)。在一小队列8例复发/难治性淋巴瘤患者中,7例达到CR,显示出初步疗效。
总之,CRISPR递送的CAR-T细胞具有精确靶向和同时敲除有害基因等优势。然而,其长期疗效和安全性需要在更大患者群体(包括B-ALL患者)中进一步验证。
上述基因敲除/敲入策略限于已知常见靶点,而CRISPR筛选为探索T细胞内的复杂调控网络提供了强大工具。多项研究使用CRISPR敲除筛选鉴定T细胞抗肿瘤功能的关键调控因子。敲除这些靶点增强了肿瘤小鼠模型(包括B-ALL)中CAR-T细胞的抗肿瘤功能,显示出临床潜力;然而临床转化还需要进一步验证。
与T细胞/CAR-T筛选相比,B-ALL原始细胞的CRISPR筛选更具疾病特异性。B-ALL原始细胞的CRISPR筛选可鉴定在免疫压力下介导CAR-T治疗敏感性或耐药性的因素。在B-ALL中,死亡受体通路基因、CD58(损害免疫突触形成)或促凋亡蛋白NOXA的敲除导致耐药。Tang等鉴定出自噬作为耐药机制。自噬相关基因的敲除增强了CAR-T杀伤,这些基因的表达水平区分了应答者和非应答者(ROC=0.776)。靶抗原表达的关键调控因子也被揭示:SPPL3敲除导致CD19过度糖基化,阻碍CAR识别,而NUDT21敲除增加CD19表达和CAR-T疗效。在B-ALL微环境中,IFNγR/JAK/STAT信号敲除通过阻断HLA-E上调使B-ALL细胞对CAR-T治疗敏感,否则HLA-E上调会通过CAR-T细胞上的NKG2A/CD94传递抑制信号。这些使用CRISPR筛选的研究揭示了影响B-ALL对CAR-T治疗反应的各种因素。未来工作应进一步验证这些分子是否可作为预测B-ALL患者CAR-T治疗疗效的生物标志物,开发靶向这些分子的药物,并研究其与CAR-T治疗联合用于耐药疾病的疗效。
优化细胞来源和CAR-T制造
为解决CAR-T可及性相关的挑战,已在优化细胞来源和制造过程方面做出重大努力,旨在为既往不符合条件的患者提供输注机会、缩短静脉到静脉时间并降低经济负担。最新发展包括异基因CAR-T产品、快速制造平台、即时生产和体内CAR-T生成(图3)。
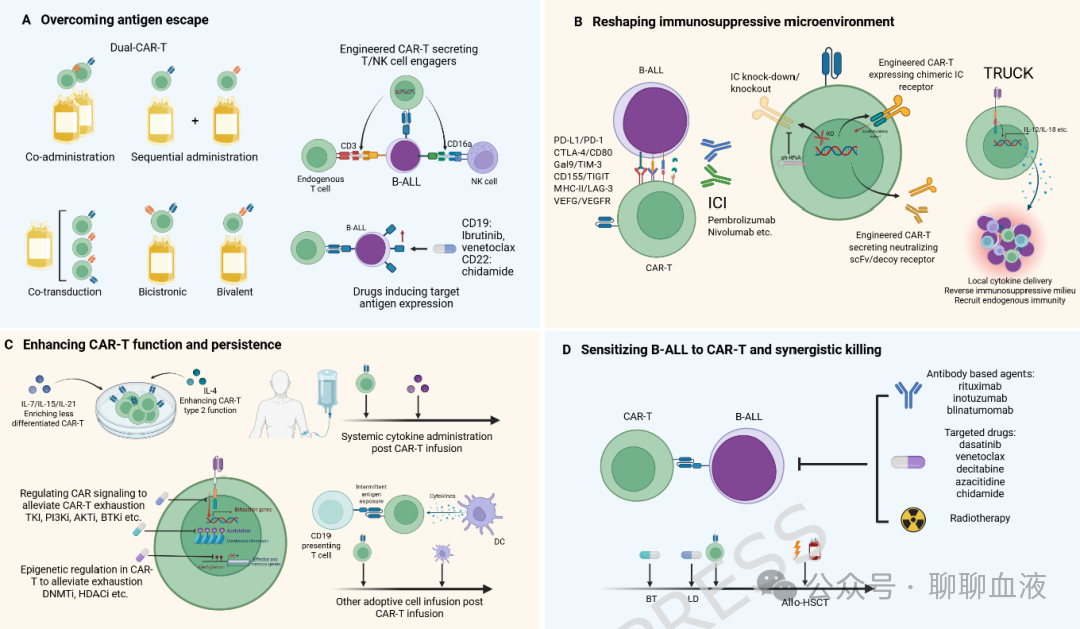
图4提高 B-ALL中 CAR-T疗效的联合策略。A:克服抗原逃逸;B:重塑免疫抑制微环境;C:增强 CAR-T细胞功能和持久性;D:使 B-ALL对 CAR-T治疗敏感并协同杀伤
供者来源CAR-T细胞主要用于异基因造血干细胞移植(allo-HSCT)后复发的B-ALL患者。因为这些患者先前已重建了供者的免疫系统,排斥供者来源CAR-T细胞的风险降低。在del Bufalo等的研究中,13例患者接受了HLA匹配或半相合供者的CAR-T细胞;所有患者均达到MRD阴性CR,8例在中位随访12个月时维持缓解。Zhang等用供者来源CD19 CAR-T治疗了43例移植后患者:34例(79%)达到完全缓解(CR),1年无事件生存(EFS)和OS率为43%,仅2例患者出现≤2级急性移植物抗宿主病(GvHD)。Roddie等报告了93%的MRD阴性CR率(13/14),并进一步证实供者CAR-T输注前的淋巴细胞清除改善了1年EFS和OS。
供者来源CAR-T细胞仍是患者特异性的。相比之下,来自第三方供者的UCAR-T提供了现货型可用性。UCAR-T细胞使用基因编辑来破坏HLA匹配要求,是R/R B-ALL研究的主要焦点。
在Benjamin等的研究中,使用TALEN敲除TRAC和CD52,21例R/R B-ALL患者接受了UCAR-T,CR率为67%(14/21),10例达到MRD阴性缓解。然而UCAR-T持久性欠佳:71%的患者接受了allo-HSCT,数据截止时仅5例存活。UCAR-T为不符合自体CAR-T条件的患者提供了一种替代方案,具有中等疗效和安全性。然而其安全性担忧包括alemtuzumab相关感染风险、潜在GvHD和脱靶毒性。疗效受限于扩增和持久性差,可能由于宿主免疫排斥,T细胞受体(TCR)敲除可能进一步损害持久性。
最近,Wu等鉴定出SPPL3敲除作为一种新策略。SPPL3敲除可增加TCR/CD3糖基化,保留TCR表达同时降低对异基因HLA分子的识别,从而在不损害CAR-T效应功能的情况下降低GvHD风险。重要的是,TCR信号对CAR-T细胞的长期体内持久性和肿瘤控制至关重要,为其TCR保留策略提供了依据。在一项初步研究中,3例患者(包括1例B-ALL)接受来自HLA不匹配供者的SPPL3敲除UCAR-T,不良事件可控,未发生典型GvHD,B-ALL患者达到MRD阴性CRi,CAR-T扩增和持久性良好。
除UCAR-T生产的基因编辑策略外,还存在非基因编辑策略。例如,工程化CAR-T细胞表达带有KDEL标签的抗CD3-scFv可阻止TCR/CD3从内质网分泌,从而降低表面TCR表达。这种非基因编辑UCAR-T治疗在8例R/R B-ALL患者中进行了初步测试,7例达到MRD阴性CR/CRi。另一种制造UCAR-T的非基因编辑策略为使用病毒特异性T细胞构建CAR-T细胞。与常规CD3激活T细胞不同,病毒特异性T细胞主要通过其TCR识别病毒抗原表位-HLA复合物,从而降低其同种反应性和GvHD风险。最近报告了一项现成的Epstein-Barr病毒特异性CAR-T细胞1期研究的结果。在6例R/R B-ALL患者中,3例达到或维持CR。然而,该研究中CAR-T细胞的持久性差,可能与制造产品中终末分化T细胞的富集有关。
除常用的αβ T细胞外,由于其固有的抗肿瘤活性和低GvHD风险,某些特殊T细胞亚群也是优化UCAR-T治疗的候选来源。一种独特的T细胞亚群是γδ T细胞,其以MHC非限制性方式识别抗原。这一特征赋予其在肿瘤识别/清除方面的天然优势以及降低的GvHD风险。类似地,不变自然杀伤T(iNKT)细胞是另一种罕见的T细胞亚型,其特征为表达不变TCR。一项使用异基因CAR-iNKT细胞治疗B细胞恶性肿瘤的研究中,9例患者中有3例达到CR/CRi。γδ T和iNKT细胞作为现成CAR-T产品的低GvHD风险可降低对基因编辑的依赖。然而,这些T细胞亚群的稀缺性在收集足够数量细胞方面构成挑战。
与有限的特殊T细胞亚群相比,脐带血(UCB)可为UCAR-T制造提供更丰富的来源。使用UCB作为T细胞来源具有多项优势:HLA匹配要求较不严格,增加通用性;低免疫原性和降低的GvHD风险;以及UCB T细胞早期分化表型的富集,赋予更强的增殖能力和延迟耗竭特征。多项临床前研究利用供者UCB制备CAR-T细胞,在B-ALL模型中显示出抗肿瘤活性。
此外,诱导多能干细胞(iPSCs)可作为CAR-T细胞原材料的重要来源。iPSCs具有自我更新能力,确保细胞供应。在iPSCs中进行的基因操作(如CAR基因插入和TRAC敲除)可在iPSC增殖过程中传递给子代细胞,避免重复编辑操作并增强细胞均一性。这种方法显示出临床应用前景。
为加速生产,Gracell Therapeutics开发了快速CAR-T(F-CAR-T),在一天内完成T细胞分离、激活和转导,省略扩增步骤。在一项25例R/R B-ALL患者的研究中,从单采到输注的中位时间为14天。所有25例达到CR/CRi,在接受巩固性allo-HSCT的20例患者中,15例(75%)在中位随访693天时达到MRD阴性CR。一项最近的回顾性研究揭示,与传统CAR-T相比,F-CAR-T导致显著更高的CR和MRD阴性率,但1年或2年无白血病生存(LFS)/OS无差异。然而,F-CAR-T与更高的CRS(91.3% vs. 66.7%)和ICANS(30.4% vs. 9.5%)发生率相关。这些发现表明,跳过体外扩增减少了细胞产量,但能够快速生产体内扩增良好的低分化CAR-T细胞,在低剂量(104-105细胞/kg)下实现高缓解率。然而,激活后的冷冻保存可能使这些细胞过度活跃,导致毒性增加。因此,快速制造的CAR-T细胞治疗需要更密切地监测不良事件,其长期疗效需要进一步评估。
POC CAR-T细胞构成另一种缩短制造时间和降低成本的策略。与传统制造需要长途细胞运输和冷冻保存不同,POC CAR-T在治疗现场使用自动化系统生产新鲜、非冷冻保存的产品,减少等待时间和成本。在Maschan等的研究中, CD19 CAR-T细胞在CliniMACS Prodigy平台上于当地站点为54例B细胞恶性肿瘤患者制造。从单采到输注的中位时间为13天。在27例可评估的R/R B-ALL患者中,24例(89%)达到MRD阴性CR。对于应答者,1年无进展生存(PFS)和OS分别为37.5%和79.2%,中位缓解持续时间(DOR)为310天。印度的VELCART试验在同一平台上治疗了10例患者(6例R/R B-ALL)。静脉到静脉时间为9天;所有6例B-ALL患者达到CR,数据截止时4例存活。因此,POC CAR-T细胞为B-ALL提供了节省时间和成本的平台。然而,广泛采用面临挑战,包括跨站点的统一质控、协调的监管框架以及大量投资需求。
体内CAR-T构成一种新兴策略,系统性递送靶向T细胞的CAR编码载体(如病毒载体或纳米颗粒),使CAR在患者体内从头表达。这种方法绕过体外激活和扩增,显著减少制造时间。载体可预先生产并储存,允许立即给药。体内CAR-T细胞也可能经历更生理性的激活,潜在减少耗竭。ESO-T01是一种体内慢病毒载体,编码B细胞成熟抗原CAR并通过TCR特异性纳米抗体靶向T细胞,在两项独立研究中进行了评估。在一项4例R/R多发性骨髓瘤(MM)患者的病例系列中,ORR为100%(2例CR,2例部分缓解),3例患者出现3级CRS。在一项5例R/R MM患者的1期试验中,ORR为80%(3例CR,1例PR)。两组的不良事件均可控,CAR-T扩增动力学与商业产品相当。这些初步MM结果验证了体内CAR-T方法的可行性,并突出了其应用于其他疾病(如B-ALL)的潜力。未来需要更大队列和更长随访期的研究来探索体内CAR-T治疗在B-ALL和其他适应症中的疗效和安全性。
增强B-ALL中CAR-T疗效的联合策略
尽管CAR-T治疗在R/R B-ALL中的初始疗效令人印象深刻,但由于抗原逃逸、CAR-T持久性有限和免疫抑制微环境,复发仍是主要挑战。为解决这些挑战,已开发联合策略以增强CAR-T功能、拓宽抗原识别或重塑肿瘤微环境。本节综述双靶点CAR-T、免疫检查点抑制剂、小分子抑制剂和细胞因子装甲策略的最新进展(图4)。
双靶点CAR-T
双靶点CAR-T细胞识别两种抗原以防止单抗原丢失导致的复发。在R/R B-ALL中,CD19-CD22是最常见的方法,策略包括两种CAR-T产品的序贯或联合给药或工程化双靶点CAR-T细胞(共转导、双顺反子或双价载体)。
目前尚无最佳策略的共识。一项回顾性研究揭示,串联治疗和序贯治疗在缓解或2年OS方面无显著差异,尽管1年LFS更利于串联治疗(71.1% vs. 52.2%,P=0.154)然而串联CD19/CD22 CAR-T细胞对CD22抗原的识别效率相对较差,可能由于串联scFv之间的空间位阻。使用共转导CAR-T或双顺反子CAR-T可增加CD22识别并防止CD22下调介导的逃逸,但这些策略增加了每个CAR-T细胞表达的CAR分子数量,可能导致激活诱导的细胞死亡或耗竭。共刺激域的选择也很重要:一项临床前研究显示,CD19.BBζ-CD22.28ζ配对产生最持久的疗效,因为CD28增强不稳定的CD22识别,而4-1BB促进稳定CD19 CAR的持久性。
总体而言,尽管靶向额外抗原,但双靶点治疗似乎未显著增加治疗相关不良事件。治疗失败主要由于CD19+CD22+复发(持久性不足),其次为CD19-CD22+复发(CD22靶向不足)。未来努力应聚焦于改善持久性和减少抗原阴性复发。除CD22外,还有多种替代靶点可用,包括自然杀伤组2成员D配体、B细胞激活因子受体和FMS样酪氨酸激酶3。这些CD19-X双靶点CAR-T构建体在临床前研究中显示出强大的抗B-ALL活性,但其疗效需要临床试验进一步验证。
免疫检查点抑制剂
CAR-T细胞治疗与PD-1/PD-L1抑制剂联合用于R/R B-ALL的临床数据有限。在一项对4例CAR-T反应或扩增不佳儿童的研究中,帕博利珠单抗延长了CAR-T持久性总体,并在2例儿童中诱导了客观反应。在另一项对13例CAR-T反应不佳儿童的研究中,PD-1抑制剂输注在6例早期B细胞恢复的儿童中恢复了3例的B细胞再生障碍。在4例髓外疾病患者中,2例达到CR,2例达到PR。除PD-1外,CTLA-4敲除增强CAR-T抗B-ALL活性, V-domain免疫球蛋白T细胞活化抑制因子的抑制在小鼠模型中与CAR-T活性协同作用。此外,最近一项研究工程化CAR-T细胞分泌T细胞免疫球蛋白和粘蛋白域蛋白3(TIM-3)-Fc诱饵受体,保护免受TIM-3/Galectin-9介导的耗竭,改善B-ALL中的抗肿瘤活性和持久性。
小分子抑制剂
达沙替尼可逆性阻断CAR/TCR信号,防止耗竭并通过表观遗传重编程恢复耗竭CAR-T细胞的功能。间歇性达沙替尼增强CAR-T治疗的抗肿瘤活性并延长B-ALL小鼠模型的生存。临床上,达沙替尼可作为Ph+ B-ALL的桥接或巩固治疗。在最近一项新诊断Ph+ B-ALL患者的研究中,达沙替尼为基础的诱导后序贯CD19/CD22 CAR-T细胞治疗实现了85%(23/27)的分子CR和92%的2年LFS/OS。
此外,PI3K/AKT/mTOR通路调控T细胞增殖和分化。在制造过程中用PI3K抑制剂和AKT抑制剂预处理CAR-T细胞已在临床前研究中显示可减少CAR-T耗竭,增加低分化表型数量,从而增强体内扩增和抗肿瘤功能。
伊布替尼是一种布鲁顿酪氨酸激酶抑制剂(BTKi),也抑制参与T细胞分化的IL-2诱导T细胞激酶。在CAR-T制造过程中加入伊布替尼增加低分化表型细胞数量并减少耗竭标志物表达。在B-ALL小鼠模型中,与单独CAR-T治疗相比,伊布替尼联合CAR-T治疗降低肿瘤负荷并延长生存。[此外,伊布替尼抑制转录重编程,使B-ALL原始细胞维持低CD19表达,从而增加CAR-T细胞细胞毒性。此外,CD19阴性B-ALL细胞变得依赖BCR-BTK-MEK通路并对BTKi敏感,提示伊布替尼与CAR-T联合可能有助于克服CD19抗原逃逸。
地西他滨可改变CAR-T细胞中与耗竭相关的DNA甲基化。在体外CAR-T细胞培养中加入低剂量地西他滨(10 nM)增加增殖和记忆相关基因表达,减少耗竭相关基因表达,并减少终末分化。在B-ALL和淋巴瘤小鼠模型中,地西他滨预处理的CAR-T细胞表现出增强的抗肿瘤活性、持久性和记忆表型维持。与临床前发现一致,一项在R/R B-ALL患者中地西他滨加入环磷酰胺(Cy)和氟达拉滨(Flu)(Cy/Flu)淋巴细胞清除方案的研究显示,地西他滨 + Cy/Flu组相比传统淋巴细胞清除组改善了长期生存。
组蛋白去乙酰化酶抑制剂(HDACi)可直接改善CAR-T功能。I类HDACi 西达本胺重塑CAR-T表观遗传学并激活Wnt/β-catenin通路,维持记忆表型并缓解耗竭。在B-ALL小鼠模型中,CAR-T预处理西达本胺或CAR-T注射后口服HDACi增强抗肿瘤功能,保留记忆表型并延长生存。此外,西达本胺通过上调CD22表达和上调促凋亡蛋白NOXA表达使B-ALL细胞对CAR-T敏感,从而增加CAR-T疗效。临床上,西达本胺作为19例CAR-T治疗后达到缓解的R/R B-ALL患者的维持治疗,在未接受allo-HSCT的13例患者中实现62.9%的1年无病生存和83.3%的1年OS。
BH3模拟物(如维奈克拉)预处理上调B细胞肿瘤上CD19和促凋亡蛋白BAK的表达,增加CAR-T细胞毒性。然而CAR-T细胞治疗期间或之后 同时使用可促进CAR-T凋亡和耗竭,限制疗效。为解决这一问题,工程化过表达抗凋亡蛋白Bcl-xL的CAR-T细胞被证明可抵抗BH3模拟物诱导的毒性,维奈克拉与Bcl-xL过表达CAR-T联合延长B-ALL小鼠模型的生存。在最近一项新诊断高危Ph阴性B-ALL患者的临床研究中,维奈克拉加阿扎胞苷诱导后CD19/CD22串联CAR-T治疗实现91.7%(11/12)的CR率,10%的MRD阴性结局。1年累积复发发生率(CIR)、LFS和OS分别为18.2%、81.8%和81.8%。
细胞因子
白细胞介素(IL)-2通常在CAR-T制造过程中添加以增加体外扩增,但它也可促进CAR-T终末分化和Treg扩增。与使用IL-2相比,制造过程中使用IL-7、IL-15或IL-21等细胞因子也促进CAR-T扩增并导致更少分化表型的发展。此外,CAR-T生成过程中添加IL-4增强2型免疫功能。具有高2型功能的CAR-T细胞在B-ALL小鼠模型中显示出显著更大的扩增、更低的耗竭和更长的生存,优于低2型功能者。
正在探索输注后的系统性细胞因子补充。在一项CD19/CD22 CAR-T联合IL-15受体激动剂NKTR-255治疗9例R/R B-ALL患者的1期研究中,89%达到MRD阴性CR/CRi,1年PFS为67%(对照组38%),仅4例患者出现1级CRS。
除全身给药外,第四代CAR-T治疗,也称为TRUCK(T细胞重定向用于通用细胞因子介导杀伤),涉及基因修饰CAR-T使其能够局部释放促炎细胞因子,以自分泌或旁分泌方式改善肿瘤微环境。CD19 CAR-T介导的IL-18分泌在淋巴瘤患者中,既往CD19 CAR-T治疗仅实现81%(17/21)的ORR和52%(11/21)的CR率,安全性特征与传统CAR-T治疗相似。另一种策略涉及工程化CAR-T细胞共表达细胞因子;最近一项研究报告CD19 CAR-T细胞表达IL-10在12例R/R B-ALL患者中实现100%的1个月总缓解率和91%/100%的6个月RFS/OS率。这些研究为细胞因子装甲CAR-T在癌症治疗中的可行性提供了临床概念验证,提示了下一代CAR-T开发的新方向。涉及先进CAR-T的试验目前正在B-ALL和其他血液肿瘤及实体瘤中进行,等待对其疗效和安全性的进一步确认。
B-ALL中围CAR-T期管理的进展
CAR-T治疗的成功实施也依赖于一系列对治疗结局有重要影响的围输注管理步骤。如图5所示,R/R B-ALL患者接受CD19 CAR-T治疗的临床工作流程包括桥接治疗、淋巴细胞清除预处理、输注后监测、巩固和挽救策略。本节讨论这些围CAR-T管理步骤中的最新进展和关键考虑因素。
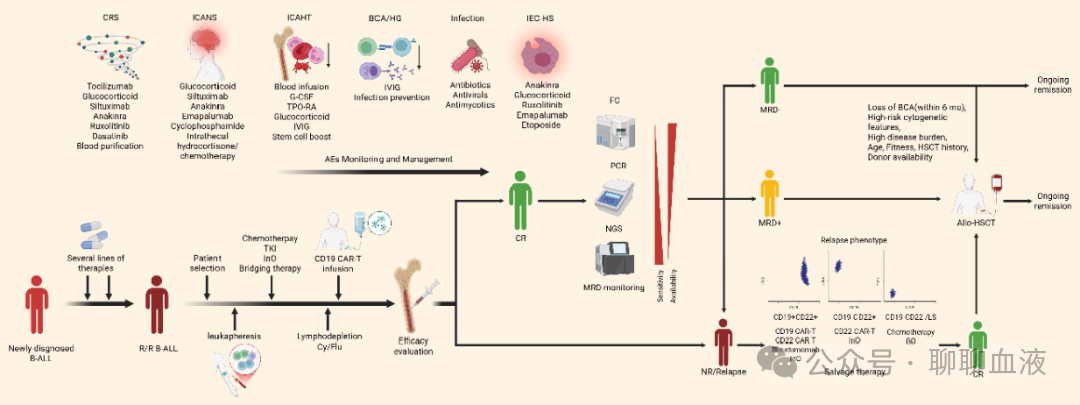
图5.接受CD19 CAR-T治疗的 R/R B-ALL患者的管理工作流程
桥接治疗和淋巴细胞清除
桥接治疗用于在CAR-T制造窗口期间控制疾病负荷。除常规化疗外,靶向治疗和免疫治疗也可作为桥接选择。TKI可作为费城染色体阳性ALL(Ph+ ALL)患者的桥接方案。尽管TKI对CAR-T增殖和活性有负向调节作用,但成功的TKI和CAR-T联合方案已在B-ALL中出现。
奥加伊妥珠单抗(InO)是B-ALL患者桥接治疗的潜在选择,与化疗相比可实现更优越的肿瘤负荷降低,但它也耗竭正常CD19+ B细胞,可能损害后续CD19 CAR-T扩增。回顾性数据显示,InO暴露(作为桥接或既往治疗)与CAR-T输注后更短的无事件生存和总生存相关。然而多变量分析提示InO暴露不再是无进展生存的独立预测因子,表明观察到的差结局可能反映了更具侵袭性、耐药性患者人群的选择,而非InO对CAR-T功能的直接负面影响。鉴于桥接特异性病例的样本量小,需要前瞻性对照试验来阐明InO桥接对CAR-T的影响。
贝林妥欧单抗与CD19 CAR-T治疗共享相同靶点。既往贝林妥欧单抗暴露与一些研究中后续CAR-T治疗后较低的缓解率和较差的长期生存相关,可能由于CD19下调或CD19阴性亚克隆的选择。然而一项大规模研究揭示,仅对既往贝林妥欧单抗无反应的患者CAR-T结局较差,而贝林妥欧单抗应答者实现了与贝林妥欧单抗初治患者相似的结果,提示T细胞功能障碍或其他耐药机制也有贡献。最近一项278例R/R B-ALL患者的真实世界研究也证明,既往贝林妥欧单抗无反应与brexu-cel治疗后显著较差的结局相关。因此,使用贝林妥欧单抗作为CD19 CAR-T治疗的桥接需要仔细评估。
CAR-T输注前一周使用Cy/Flu进行淋巴细胞清除(LD),通过清除内源性淋巴细胞、降低肿瘤负荷、促进稳态细胞因子和减轻抗CAR免疫来增强CAR-T扩增和持久性。然而LD的最佳剂量不固定;临床试验中常用剂量为总Flu浓度75-120 mg/m²和总Cy浓度750-1500 mg/m²,通常分1-5天给予。值得注意的是,更高的Flu暴露(AUC >13.8-14 mg·h/L)与更好的结局和更低的CD19阳性复发相关,而较低的暴露预测更短的生存。鉴于Flu药代动力学的个体间差异,固定剂量并非最佳。最近一项研究提出了年龄调整或药代动力学监测剂量(以第1天Flu水平为指导),实现了更高的目标暴露、改善的总生存和降低的复发率。
总之,现有证据表明LD方案和剂量的选择以及药代动力学监测显著影响R/R B-ALL CAR-T治疗的疗效。然而在优化LD方案以实现最佳疗效的同时,也应权衡不良效应如骨髓抑制的潜在风险。
也探索了替代LD方案。在CD19/CD22 CAR-T前将地西他滨加入Cy/Flu未改善早期MRD阴性,但显著改善了3年LFS(92.9% vs. 27.3%)和OS(92.3% vs. 41.7%)。一例儿童患者克拉屈滨联合Cy的病例报告显示未损害后续CAR-T疗效。对于异基因CD52敲除CAR-T,在Cy/Flu中加入alemtuzumab增强CAR-T扩增和反应率,但增加骨髓抑制和感染风险。尽管淋巴细胞清除是CAR-T治疗的标准步骤,其具体实施(包括药物组合、剂量和时机)需要进一步深入研究,以基于B-ALL患者的异质性建立个体化LD策略。最后,新兴的体内CAR-T制造可能减少对LD的依赖,潜在重新定义其未来作用。
监测和巩固治疗
输注后动态监测对早期复发检测至关重要,输注后可监测CAR拷贝数。低峰值CAR扩增或第0-28天的低AUC与抗原阳性复发相关,但其不反映CAR-T功能状态,更常见的是监测外周血B细胞水平。CD19 CAR-T介导的B细胞再生障碍(BCA)反映CAR-T细胞在体内的功能持久性,而B细胞恢复(BCR)提示持久性丧失。已建立BCA的国际共识定义以标准化输注后监测。根据该共识,BCA定义为骨髓中<0.01% CD19+ B细胞(占总白细胞或单个核细胞)和外周血中0 CD19+ B细胞/μL。然而需注意,部分患者在复发时可能仍有持续的B细胞再生障碍,表明监测BCA的局限性。
MRD是预测B-ALL复发的另一个重要生物标志物,可通过流式细胞术、定量聚合酶链反应或下一代测序(NGS)检测。NGS提供极高的敏感性(10-6),能够在形态学复发前早期检测MRD,从而为后续干预提供更多时间。研究揭示,3个月时骨髓样本中NGS-MRD阳性的检测与B-ALL复发强烈相关。推荐结合外周血B细胞监测与MRD评估,以更准确地识别疾病复发。
Allo-HSCT是CAR-T诱导缓解后的关键巩固策略,但关于CAR-T细胞诱导缓解后桥接allo-HSCT是否延长R/R B-ALL患者生存的报道存在矛盾。
因此,识别哪些类型的R/R B-ALL患者更适合并可能从桥接allo-HSCT中获益至关重要,而其余患者可能通过密切监测或其他低毒性巩固策略在CAR-T治疗后实现长期生存。当移植可行时,应首先识别具有复发高危特征的患者。MRD状态是复发的最重要预测因子之一,对于达到MRD阳性缓解的患者应考虑巩固性allo-HSCT。在Gu等的研究中,巩固性allo-HSCT与MRD阳性缓解患者的LFS和OS改善相关。早期BCR是另一个危险因素:Summers等报告对于63天内BCR的患者,allo-HSCT改善了LFS。高危遗传学也指导决策:Zhang等显示在TP53+患者中,allo-HSCT改善了1年LFS和1年OS。类似地,Jiang等报告输注前低MRD(<5%)但细胞遗传学不利或高肿瘤负荷(≥5%)的患者从allo-HSCT中获益,EFS和OS均改善。
除复发危险因素外,其他因素也会影响allo-HSCT决策。在ELIANA试验中,达到tisa-cel CR且未接受allo-HSCT的儿童/年轻成人患者2年和3年RFS率分别为81%和76%,而接受移植治疗或其他巩固的患者3年RFS率为48%(剔除巩固后为52%)。真实世界CIBMTR数据证实儿童患者可单独通过CAR-T治疗实现持久缓解(中位RFS 36个月),提示在该人群中将CAR-T作为确定性治疗是可行的。相比之下,大多数研究表明成人患者从CAR-T后allo-HSCT中获益,导致推荐对CAR-T治疗后达到缓解的成人B-ALL患者进行巩固性allo-HSCT。
CAR-T共刺激域对移植决策的影响仍不清楚。在ZUMA-4研究(接受brexu-cel治疗的儿童/年轻成人)中,大多数患者接受了allo-HSCT;剔除移植后的中位DOR为7.2个月(未剔除为14.2个月),剔除后的中位RFS为5.2个月(未剔除为7.4个月)。Shah等报告在接受CD28为基础CAR-T治疗达到CR的成人中,所有7例未移植患者复发,而21例移植患者中仅2例复发(5年EFS 61.9%),提示CD28为基础CAR-T可能需要移植来补偿其有限的持久性。既往接受过allo-HSCT的患者接受CAR-T后是否从第二次移植中获益存在争议,一项研究显示首次和第二次移植之间2年LFS或OS无差异。供者选择、时间间隔和预处理方案也可能影响移植结局。未来需要大规模随机对照试验来评估巩固性allo-HSCT的作用,以及更多亚组特异性研究来为B-ALL患者提供个体化临床决策。
除allo-HSCT外,还探索了其他巩固策略以增加CAR-T持久性。CD19抗原暴露不足与CD19 CAR-T体内持久性差相关,为解决这一问题,将CD19 CAR-T与人工表达CD19的自体T细胞(T-APCs)联合提供间歇性抗原刺激以增加CAR-T增殖和持久性。在PLAT-03试验(19例R/R B-ALL患者)中,T-APC输注未引起CRS或神经毒性,显著延长CAR-T持久性(P=0.03)。另一项在13例新诊断Ph+ B-ALL患者中的研究使用多周期CD19 CAR-T、CD19表达T细胞和TKI进行巩固治疗,实现中位DOR 27个月,RFS和OS率分别为84%和83%。
树突状细胞(DC)疫苗也可促进CAR-T扩增。在一项8例接受CD19 CAR-T后达到MRD阴性CR的R/R B-ALL患者的研究中,后续DC疫苗接种诱导CAR-T再扩增,中位持久性336天,优于历史CAR-T单药治疗。这些发现提示此类巩固策略可能为B-ALL患者提供allo-HSCT的替代方案。然而,这些创新方法需要在更大队列中进一步验证。
挽救治疗
尽管CD19 CAR-T细胞在R/R B-ALL患者中实现高缓解率,约50%的患者在CAR-T输注后复发,部分患者在首次输注后未能达到缓解。基于白血病原始细胞上的CD19表达,难治性或复发性疾病可分为抗原阳性或抗原阴性,指导不同挽救策略的选择。
CD19阴性患者的原始细胞常保留其他B细胞抗原,使替代抗原靶向成为关键挽救策略,其中CD22是最常用的靶点。InO可用于CD19阴性复发后,CD22 CAR-T治疗在此类患者中诱导约60%-80%的缓解,尽管反应常受CD22下调限制。因此,推荐CD22 CAR-T后桥接HSCT以巩固反应。
此外,几种旨在提高CAR-T对抗原识别敏感性的临床前策略可能有助于增强对低密度靶细胞的清除,尽管这些策略的疗效需要临床验证。
谱系转换是一种特殊的抗原阴性复发形式,涉及淋巴抗原的丢失,使替代淋巴抗原靶向无效。对于谱系转换的挽救治疗尚无共识。病例报告描述了gemtuzumab ozogamicin联合氟达拉滨和高剂量阿糖胞苷或达沙替尼和维奈克拉在BCR::ABL1+患者中的成功再诱导。
对于CD19阳性疾病,CD19 CAR-T细胞的再输注可用作挽救治疗。然而与初始CAR-T治疗相比,再输注诱导较低的CR率,约20-50%的B-ALL患者在相同CAR-T产品再输注后实现第二次缓解,一个潜在原因是反复输注鼠源CAR-T细胞可在患者中诱导抗CAR免疫反应。免疫原性较低的人源化CAR-T细胞已改善结局:一项研究报告CR/CRi率为79%(26/33),中位RFS 29个月。
也可使用InO或贝林妥欧单抗,与CAR-T细胞再输注相比提供更大的可及性且无制造延迟。Wudhikarn等使用贝林妥欧单抗±InO治疗7例CAR-T后抗原阳性复发的B-ALL患者,其中3例(43%)达到CR。Qi等使用贝林妥欧单抗作为5例对CD19 CAR-T治疗难治或复发B-ALL患者的挽救治疗,4例(80%)达到CR/CRi。最近一例病例报告表明,供者来源CAR-NK治疗挽救了一例CAR-T难治性儿童B-ALL患者,诱导缓解并实现HSCT。鉴于现有研究样本量小,需要更多研究来评估这些挽救策略在CD19阳性疾病患者中的疗效和安全性。
将CAR-T推向B-ALL早线治疗
鉴于CAR-T在R/R B-ALL中的强大抗白血病活性,将CAR-T推向更早期治疗线是重要趋势。目前接受CAR-T的患者通常已接受多线既往治疗,广泛的既往治疗损害骨髓储备和T细胞质量,与不良结局相关。早期应用(如首次复发或初诊时)可减少累积化疗毒性、简化治疗并改善长期预后,特别是对于高危患者。在较低疾病负荷时使用CAR-T(如缓解后巩固或MRD根除)也可减轻CRS/ICANS风险、实现更深清除并可能减少后续治疗需求。
CAR-T细胞已被探索作为新诊断B-ALL的一线治疗。Zhang等用达沙替尼为基础诱导后序贯CD19/CD22 CAR-T治疗新诊断Ph+ B-ALL成人患者。在27例患者中,85%在CD19 CAR-T后达到分子CR,接受CD22 CAR-T的25例患者中76%也达到分子CR。中位随访23.9个月时,仅2例复发,2年LFS和OS均为92%。类似地,Li等在新诊断高危Ph阴性B-ALL患者中使用维奈克拉/阿扎胞苷诱导后串联CD19/CD22 CAR-T细胞。CR率为91.7%(10例MRD阴性)。中位随访17个月后,3例复发(均为CD19阳性),1年CIR、LFS和OS分别为18.2%、81.8%和81.8%。
CAR-T作为MRD阳性B-ALL的巩固治疗也在多项研究中进行了评估。Niu等治疗了15例成人患者(11例作为一线巩固,4例作为复发挽救后);所有达到MRD阴性缓解。在11例一线患者中,仅1例接受allo-HSCT,剩余非移植患者的12个月RFS和OS分别为77.8%和80.8%。Schwartz等在7例首次缓解MRD阳性成人中输注CD19 CAR-T;6例在中位3个月时达到NGS-MRD阴性。此外,在Aldoss等的研究中,首次缓解的老年(≥55岁)B-ALL患者,无论MRD状态如何,接受CAR-T作为最终巩固治疗。在11例入组患者中,治疗耐受良好。中位随访175天后,仅1例Ph+ B-ALL患者经历分子复发,其余患者维持MRD阴性缓解。这些初步数据表明,CAR-T细胞作为缓解后巩固治疗可能维持高危或MRD阳性患者的深度缓解,潜在提供移植的替代方案。
总之,CAR-T在B-ALL患者中的早期使用,特别是对于高危患者,是一种有吸引力的策略,但还需要进一步验证。关键未解决问题包括:CAR-T与其他越来越多用于一线的免疫治疗(如贝林妥欧单抗和奥加伊妥珠单抗)的最佳序贯; CAR-T巩固是否能减少后续治疗或预防CNS复发;以及限制广泛早期应用的高成本。
结论
CAR-T治疗构成了R/R B-ALL治疗的里程碑,实现了显著的CR率,为耗尽常规治疗的患者提供了强有力的治疗选择。通过CAR设计、制造过程和联合策略的持续优化,CAR-T产品已发展为增强疗效、降低毒性和改善可及性。然而若干挑战阻碍了长期成功,包括抗原逃逸复发、CAR-T细胞持久性有限以及显著毒性效应如CRS、ICANS和血液学毒性。正在研究的有前景的策略包括双靶点CAR、CRISPR工程化CAR-T、异基因CAR-T产品以及与其他药物的合理联合。此外,将CAR-T治疗整合到更早期治疗线,包括一线或首次复发设置,是最大化治愈潜力同时减轻多线治疗相关累积毒性的重要未来方向。
总之,虽然CAR-T治疗已转变B-ALL的治疗格局,其全部潜力仍在发展中。未来努力应聚焦于验证新方法以克服耐药和持久性差、改善安全性特征、优化治疗时机并拓宽可及性,以充分实现CAR-T治疗的治愈前景。
参考文献
Zha C. & Wang Y. Challenges and advances in CAR-T cell therapy for B-ALL. Biomark Res (2026). https://doi.org/10.1186/ s40364-026-00933-z