首页 > 疾病防控/ 正文
深度解析医学证据,lxfs.net为你支撑决策
Sertoli–Leydig细胞瘤(SLCTs,塞尔托利-莱迪细胞瘤/卵巢支持-间质细胞瘤)是一种罕见的卵巢肿瘤,占所有卵巢肿瘤的比例不到 0.5%。它们通常影响年轻女性,并常表现为雄激素过多症状。本文报告一例独特的病例,一名 40 岁女性同时被诊断患有SLCT和透明细胞乳头状肾肿瘤(CCP-RCC,透明细胞乳头状肾细胞癌),这是一种罕见的肿瘤关联,其发病机制尚不明确。两种肿瘤均通过手术切除。诊断性检查包括激素检测、影像学检查以及基因检测,包括DICER1突变分析和多重连接依赖性探针扩增(MLPA),以及涵盖约 280 个癌症相关基因的下一代测序(NGS)检测。组织病理学检查证实为高分化SLCT和CCP-RCC。全外显子组测序或MLPA均未发现DICER1致病性变异。扩展的NGS检测也未发现临床相关改变,因此排除了已知的遗传易感性。两种肿瘤同时发生且未发现基因组改变,可能提示为偶发事件或目前尚未明确的机制。本病例强调了多学科方法在处理罕见肿瘤复合体中的重要性。DICER1突变的排除以及遗传学发现的缺失为有限的文献增添了新的证据,并强调了长期监测以及对潜在共享致癌通路进行进一步研究的重要性。

摘自《WHO(2020)女性生殖系统肿瘤分类》

摘自《WHO(2022)泌尿系统和男性生殖器官肿瘤分类》
背 景
卵巢Sertoli–Leydig细胞瘤(SLCTs)是生殖道的罕见肿瘤,占卵巢肿瘤的比例不到 0.5%,起源于Sertoli间质细胞。SLCTs主要影响育龄期年轻女性,平均诊断年龄为 25 岁,但可发生于任何年龄,包括儿童期和绝经后。SLCTs常具有激素活性,导致血清雄激素水平升高,并且确实是最常见的男性化卵巢肿瘤。临床表现高度可变,从影像学检查中的偶然发现,到与激素产生相关的症状,如男性化、月经不规律,或由肿瘤占位效应引起的腹痛。该肿瘤通常为单侧,双侧发生罕见(<1.5%),且通常局限于卵巢。虽然大多数SLCTs为良性,手术切除后预后极佳,但部分肿瘤可能表现出恶性行为。目前已提出多种预后因素用于预测SLCTs的恶性潜能,包括肿瘤大小、组织学分级、核分裂指数以及异源性成分的存在。近期研究已将SLCTs与DICER1综合征联系起来,DICER1综合征是一种罕见的遗传性疾病,使患者易患多种肿瘤。DICER1突变,特别是影响RNase IIIb结构域的突变,已在约 60% 的SLCTs中被发现,尤其是在中低分化组织学类型的SLCTs中。相比之下,高分化SLCTs通常与DICER1改变无关,提示可能存在不同的肿瘤发生通路。鉴于这些发现的重要性,基因检测已成为SLCT患者诊断性评估中不可或缺的一部分。在精准医学时代,基因组分析在阐明罕见肿瘤的发病机制和调整治疗策略方面发挥着核心作用。然而,部分患者中可检测突变的缺乏使诊断和治疗变得复杂。基于病例的基因组研究,特别是在罕见肿瘤组合中,对于探索潜在的共同分子机制和改进随访方案具有重要价值。本文报告一例独特的病例,一名 40 岁女性被诊断为纯卵巢SLCT并同时发生透明细胞乳头状肾肿瘤,且未检测到基因组DNA改变。本病例突显了诊断挑战,以及需要考虑罕见肿瘤关联和可能的潜在遗传易感性。同时,它也强调了包括激素、影像学和基因检测在内的综合诊断策略对于确保有效管理和治疗的重要性。
研究材料和方法
病例展示
患者女,40 岁,因继发性闭经 12 年以及进行性多毛症(累及面部、胸部和四肢)就诊于AOU费德里科二世大学医院内分泌门诊。其既往病史包括高血压、血脂异常、II度肥胖、甲状腺肿以及双侧非分泌性肾上腺皮质腺瘤。无已知恶性肿瘤家族史。体格检查显示,患者表现出男性化体征,包括阴蒂肥大、声音低沉、脱发,以及使用改良Ferriman–Gallwey量表评分,结果为 27 分,提示重度多毛症。
血清检测
血清检测在费德里科二世大学医院临床实验室进行。皮质醇、总睾酮、雌二醇、孕酮、促黄体生成素(LH)、促卵泡激素(FSH)、促肾上腺皮质激素(ACTH)、甲胎蛋白(AFP)、糖类抗原19-9(Ca19-9)、糖类抗原15-3(Ca15-3)、糖类抗原125(Ca125)、人绒毛膜促性腺激素β亚单位(β-HCG)和癌胚抗原(CEA)水平采用化学发光免疫分析法(CLIA)检测。血清脱氢表雄酮硫酸酯(DHEAS)和雄烯二酮水平采用化学发光免疫分析法(CLIA)检测。血清17-羟孕酮(17OHP)水平采用酶联免疫吸附试验(ELISA)检测。
研究结果
诊断
激素检测(表1)显示血清睾酮(518.9 ng/dL)和雄烯二酮水平(>10 ng/mL)显著升高,提示雄激素产生性肿瘤活性。硫酸脱氢表雄酮(DHEAS)和17-羟孕酮(17-OHP)水平在正常范围内,排除了肾上腺源性雄激素过多。
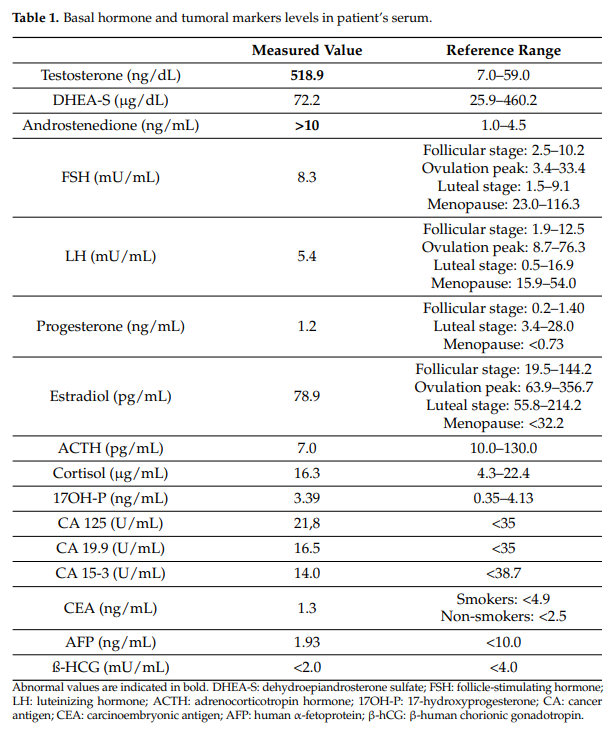
表1 患者血清中的基础激素和肿瘤标志物水平
肿瘤标志物CA 125、CA 19-9、CA 15-3、癌胚抗原(CEA)、甲胎蛋白(AFP)和β-人绒毛膜促性腺激素(β-hCG)的血清水平均在正常范围内(表1)。在激素检测之后,影像学检查对于进一步明确附件肿块的特性至关重要。超声检查因其安全性、诊断效能以及在由妇科肿瘤专业培训人员操作时能够早期提示可疑病变的优势,仍然是评估卵巢病变的首选方法。Sertoli–Leydig细胞瘤(SLCTs)虽然罕见,但通常表现为单侧实性或实性–囊性附件病变,边界清晰,多普勒检查可见丰富血流信号。虽然这些特征并非特异性,但有助于将SLCTs与更常见的卵巢上皮性肿瘤相鉴别。对于无法进行经阴道超声检查的患者,经直肠扫描可能是改善盆腔显像的有效替代方案。然而,患者拒绝接受经阴道和经直肠超声检查,因此仅能进行经腹盆腔超声检查。由于患者的体型,其诊断效能有限,因此进行了腹部和盆腔磁共振成像(MRI)。MRI显示左侧卵巢有一个 57×50 mm的不均质肿块,伴有对比增强。此外,在右肾上极发现一个最大径达 44 mm的复杂性囊性病变。
为评估肾上腺功能,进行了250 µg促肾上腺皮质激素(ACTH)肌内注射刺激试验。肾上腺对ACTH试验的反应正常,皮质醇和17-羟孕酮水平在预期范围内升高。DHEA-S水平在 30 分钟和 60 分钟后仅轻微升高。最后,睾酮和雄烯二酮浓度在试验开始时及试验过程中均呈病理性升高。皮质醇和17-羟孕酮的正常反应证实了肾上腺功能完整。然而,减弱的DHEA-S反应,加上持续高水平的睾酮和雄烯二酮,提示高雄激素血症的非肾上腺来源,强烈提示为卵巢来源(表2)。
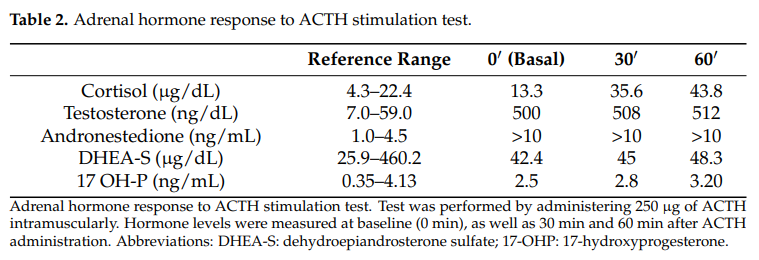
表2 肾上腺激素对ACTH刺激试验的反应
随后,进行了促性腺激素释放激素(GnRH)刺激试验,皮下注射 0.1 mg布舍瑞林,试验前于晚上 11 点口服 1 mg地塞米松抑制肾上腺功能,以在排除肾上腺雄激素影响的情况下评估卵巢功能。结果如表3所示。尽管地塞米松抑制了肾上腺雄激素,但患者在GnRH刺激后雄烯二酮反应显著升高。这一发现证实了卵巢对促性腺激素的反应性,并提示存在一个具有类固醇生成活性的卵巢肿瘤。
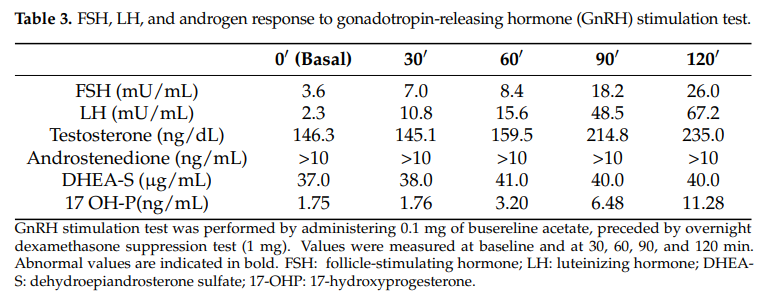
表3 FSH、LH和雄激素对促性腺激素释放激素(GnRH) 刺激试验的反应
手术治疗
患者接受了机器人辅助腹腔镜左侧输卵管卵巢切除术。卵巢肿块的组织病理学检查证实为高分化SLCT,大小为 50×43×36 mm,呈实性囊性结构,外观呈出血性棕色,占据了整个卵巢。显微镜下,肿瘤由中等大小细胞组成,核圆形,胞浆丰富嗜酸性,透明细胞极少。未见包膜侵犯。免疫组织化学分析显示Calretinin、WT1、Inhibin阳性,MART-1局灶阳性,Ki67增殖指数为 7%。在同一手术中,患者接受了机器人腹腔镜右肾部分切除术(图1b)。组织病理学分析将病变确定为CCP-RCC,2级。肿瘤大小为 47×45×31 mm,边界清晰,由乳头状结构和透明细胞巢组成,细胞核呈多形性。免疫组织化学染色CK7阳性,CD10呈"斑片状"阳性,GATA3染色阴性。虽存在局灶性坏死,但未见包膜侵犯。
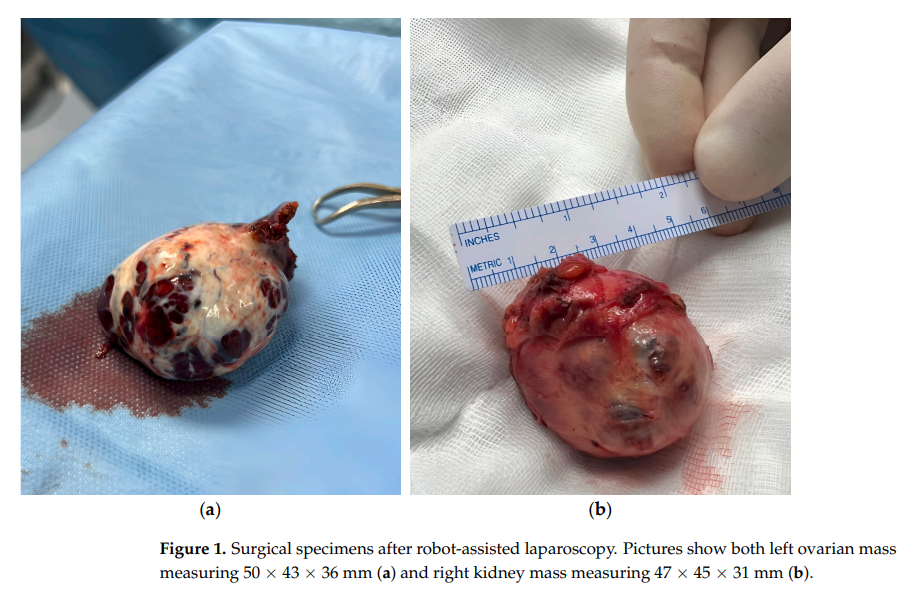
图1 机器人辅助腹腔镜手术后的手术标本
基因研究
鉴于SLCT与DICER1综合征的关联,进行了基因检测以验证DICER1基因是否存在突变。使用核酸提取试剂盒从患者外周血中提取基因组DNA。全外显子组测序(WES)平均测序深度至少为 100×。作为对该分析的补充,进行了多重连接依赖性探针扩增(MLPA)检测,以检测DICER1基因内的任何缺失或重复。WES和MLPA分析均未发现DICER1基因的点突变、移码突变和拷贝数变异,排除了DICER1相关的遗传易感性。此外,使用高通量下一代测序(NGS)对约 280 个可能与遗传性和散发性癌症相关的基因进行了突变筛查。该方法可在 8–15% 的病例中识别出致病性或可能致病性突变,并提高了下一代测序在肿瘤学中的诊断、预后和治疗价值。在所分析的其他基因中未检测到任何突变,仅在MSH6基因(NM_000179.2)中发现一个变异(c.1814C>G p.(Thr605Ser, rs587781616),外显子:4/10)。MSH6被列为林奇综合征和家族性子宫内膜癌的致病基因,两者均为常染色体显性遗传疾病。该变异存在于dbSNP数据库中,并在ClinVar中被注释为致病性解读存在冲突的变异,其中 9/10 次被描述为林奇综合征和遗传性癌症易感性的意义未明变异(VUS)。基因检测方法、其结果及临床相关性的概述总结于表4。
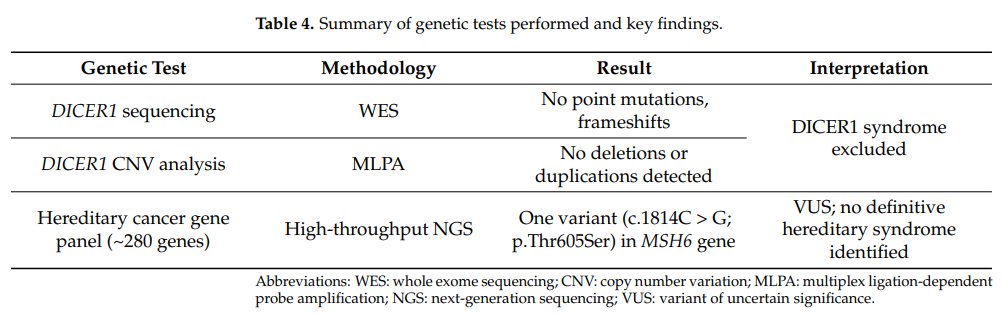
表4 所进行的基因检测及主要结果汇总
讨 论
这一病例凸显了SLCTs在诊断和治疗上的挑战,尤其是当其与其他罕见肿瘤如CCP-RCC同时发生时。虽然SLCTs罕见,但对于出现男性化症状的女性应予以考虑。本例患者表现出典型的雄激素过多体征,从而启动了全面的诊断评估。初始影像学检查发现卵巢和肾脏均有病变。诊断经组织病理学证实,这仍然是诊断卵巢肿瘤的金标准。机器人腹腔镜入路实现了肿瘤的完整切除,这对于诊断和获得最佳治疗效果均至关重要,因为SLCTs可能具有恶性潜能。
CCP-RCC的同步诊断引人深思,并引发了关于可能存在遗传关联的疑问。遗传性乳腺癌和卵巢癌综合征以及林奇综合征是与妇科肿瘤相关的最常见遗传性综合征,但其他形式的遗传性癌症也可能发生。这些形式通常与遗传性基因突变相关,临床上检测到肿瘤关联具有重要的临床意义,因为它可能有助于揭示尚未被诊断的综合征,并建议患者进行遗传咨询和正式的风险评估。SLCTs和肾脏肿瘤最近已被认为与DICER1综合征相关,DICER1综合征是一种遗传性癌症易感综合征,也称为DICER1相关性胸膜肺母细胞瘤癌症易感综合征(OMIM 601200)。DICER1基因(OMIM 606241)位于14q32.13(hg18),由27个外显子组成。它编码胞质内切核糖核酸酶(RNase)III,该酶在RNA干扰通路中发挥核心作用。它将双链RNA分子切割成小RNA,包括微小RNA(miRNA)和小干扰RNA(siRNA)。DICER1促进这些RNA整合到Argonoute蛋白中,形成RNA诱导沉默复合体(RISC)。活化的RISC识别特定的mRNA靶序列,并可启动分子降解或抑制其翻译。DICER1综合征(OMIM#601200)是一种罕见的多效性肿瘤易感综合征,以常染色体显性方式遗传,但也可在胚系中以新生突变或体细胞嵌合形式发生。据估计,80% 的致病性胚系变异遗传自父母一方,20% 为新发突变。此外,DICER1突变发生于多种癌症中,例如散发性胸膜肺母细胞瘤;性腺肿瘤、Wilms瘤和子宫内膜肿瘤;以及肾间变性肉瘤。伴或不伴SLCT的多结节性甲状腺肿-1(MNG1)(OMIM 138800)也由DICER1基因杂合突变引起。已报道的大多数DICER1突变为伴随功能丧失的点突变或移码突变,但也已鉴定出整个DICER1基因座的缺失以及框内或框外的DICER1基因内缺失。本例患者DICER1突变和该基因缺失检测均为阴性。与现有知识一致,本例患者的高分化SLCT中未发现DICER1突变,该亚型通常与DICER1变异无关。这一发现提示本例中的SLCT和CCP-RCC可能为偶发发生,而非遗传性癌症综合征的一部分。然而,不能排除其他遗传因素或信号通路促进了这些肿瘤发展的可能性。为了探索可能的遗传原因,研究人员还使用NGS检测了约 280 个已知与多种肿瘤相关的基因。除在MSH6基因(NM_000179.2)中发现一个此前报道的意义未明变异(VUS)外,未发现其他突变;该变异收录于dbSNP数据库,并在ClinVar中注释为致病性解读存在冲突。
本研究的一个局限性在于无法深入检测肿瘤组织中的体细胞突变。尽管胚系DNA的WES和MLPA均排除了已知的DICER1点突变和拷贝数改变,但研究人员认识到,深部内含子变异和体细胞嵌合现象(在中低分化SLCTs中尤其相关)在不进行肿瘤组织测序的情况下可能无法排除。我们意识到这一局限性,但由于技术和后勤条件的限制,无法进行必要的分析来深入探究这一方面,而这本可为进一步揭示可能驱动肿瘤发展和进展的遗传改变提供更深入的见解。此外,可能使个体易患多原发肿瘤的表观遗传改变也尚未排除。
SLCT的预后取决于肿瘤特征,如大小、分级和核分裂指数。大多数SLCTs预后极佳,但部分可能具有侵袭性。本例患者的高分化肿瘤提示预后良好;尽管如此,定期随访至关重要,尤其是在合并CCP-RCC的情况下。
结 论
本病例报告呈现了卵巢SLCT与CCP-RCC罕见的同步发生,且未检测到DICER1或其他致病性胚系突变。据研究人员所知,这是这两种肿瘤关联的首例报道。它强调了在研究多发性罕见肿瘤患者时,考虑潜在遗传易感性的重要性。尽管肾脏病变可能为偶然发现,但在一名相对年轻的成人中同步发生,提示可能存在共同的致病机制,值得进一步的分子研究。两种肿瘤手术切除后均获得良好结局,凸显了包括内分泌科、影像科、外科和病理科在内的多学科诊疗方法的价值。尽管进行了广泛的检测仍未发现基因组改变,提示可能涉及表观遗传因素、当前检测方法未能检出的体细胞突变,或尚未明确的遗传性癌症综合征。这些发现强调了对SLCTs的分子图谱及其可能关联进行进一步研究的必要性。在类似病例中,即使未检测到突变,也应考虑进行遗传咨询。最后,通过多中心登记进行更广泛的数据收集,可以提高对罕见肿瘤组合的理解,并促进新遗传因素的识别。这些努力最终可为临床监测策略和未来的诊断指南提供依据。
参考文献:
Macera, M.; Morra, S.; Ascione, M.; Terracciano, D.; Ianniello, M.; Savarese, G.; Alviggi, C.; Bifulco, G.; Longo, N.; Colao, A.; et al. Synchronous Ovarian Sertoli–Leydig Cell and Clear Cell Papillary Renal Cell Tumors: A Rare Case Without Mutations in Cancer-Associated Genes. Curr. Oncol. 2025, 32, 429. https://doi.org/10.3390/curroncol32080429
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP
