深度解析医学证据,DeepEvidence为你支撑决策
结构变异(SV)是人类基因变异的主要来源,对人类特征和疾病有着重要影响。目前检测SV的金标准是高精度长读长WGS技术,但这些技术的成本较高,阻碍了其大规模应用。而基于相对较小队列的高精度从头组装构建泛基因组图谱,将大型队列的短读长测序数据映射到该图谱进行SV基因分型是另一种检测策略,也被认为是检测SV最强大且最可靠的方法之一,这凸显了建立综合性SV参考panel面板以实现单核苷酸多态性(SNP)数据精准插补的迫切需求。
近日,西湖大学杨剑团队基于241名个体的高精度长读长WGS数据构建了482个单倍型解析基因组组装,全面识别171,233个高质量、全基因组范围的SV,并基于此开发了参考panel及高精度SV插补工具ImputeSV,利用SNP数据对鉴定的SV进行插补。基准测试显示,该SV插补方法展现出较高的召回率、精确度和基因型一致性。在英国生物样本库(UKB)参与者中,该方法插补了54,578个常见SV,量化了SV所解释的多种复杂性状变异,并对2,624个性状进行了全基因组SV关联分析(SV- GWAS)。该研究证明了利用长读长测序组装数据从SNP插补SV的有效性,揭示了SV在复杂性状变异中的作用,并提供了UKB中SV关联的完整目录。

研究团队在高覆盖度PacBio HiFi测序数据中获得了482个经单倍型解析的基因组组装,这些数据源自241名不同种族的个体。其中,每个基因组组装平均识别出15,765个高置信度的非着丝粒常染色体SV(图1)。鉴于相同的SV在不同基因组组装版本中可通过不同的表达方式被检测到,研究人员通过变异整合,最终在所有组装版本中鉴定出171,233个独特SV,中位长度为160bp,平均长度为1,589bp。
研究团队还建立了一套分析流程,通过对每个组装版本中各SV等位基因序列进行分析来识别可变数目串联重复序列(VNTR),共鉴定了18,360个常染色体VNTR位点。每个VNTR至少包含2个替代等位基因,平均每个位点有21.4个等位基因,779个位点含有100个及以上等位基因。约66.0%的SV位于串联重复区域(TRRs),长度与其他SV存在差异。为便使用,研究人员基于组装数据获得的变异信息,对标准插补流程进行了优化(ImputeSV),构建了适用于常见SV和特定VNTR的优化参考panel。
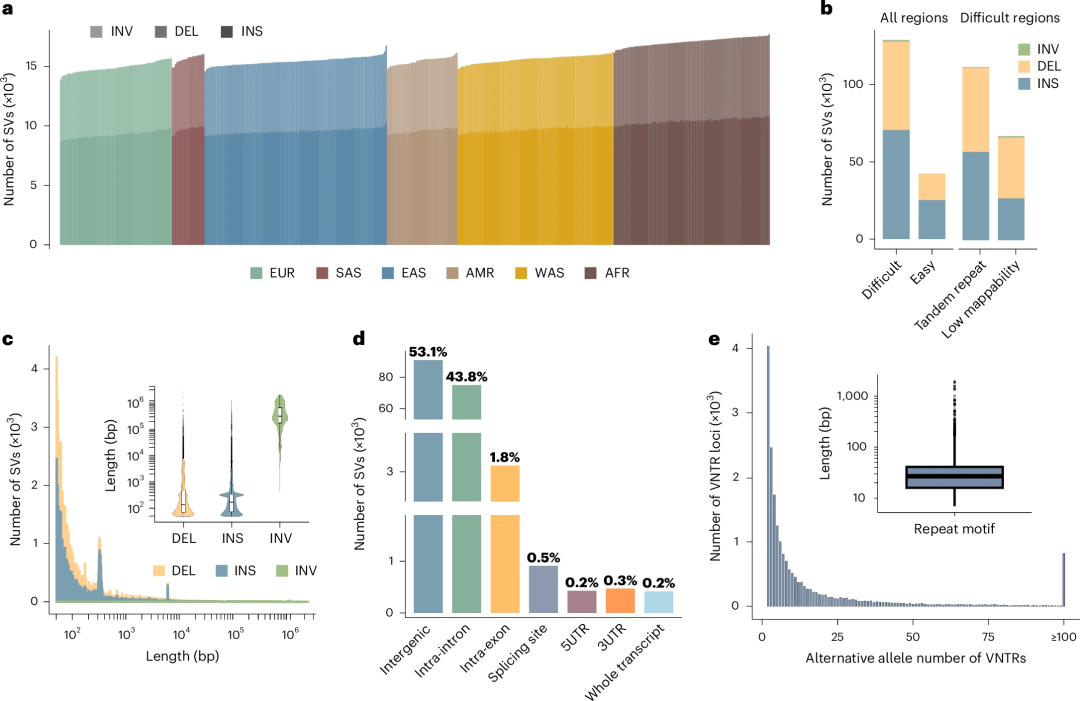
图1.在482个长读长测序中检测到的SV和VNTR。
对上述数据集进行SV插补性能评估显示,该分析流程与GIAB、HGSVC3和1KGP3等权威基准数据集相比,展现出较高的召回率、精确度及基因型一致性,在复杂且具有医学意义的基因组区域表现更突出(图2)。基于芯片数据进行SNP预插补显著提升了SV插补效果,性能几乎与使用WGS衍生SNP相当。在群体层面,常见SV在不同遗传背景下的插补精度均较高。此外,比较显示高覆盖度HiFi检测panel提供了更全面的全基因组SV覆盖度,准确插补的SV数量超过同类方法的2倍。同时,VNTR特异性检测panel成功捕捉了不同种族背景下的多等位基因VNTR长度变异,并在UKB区域与srWGS检测结果表现出高度一致性。
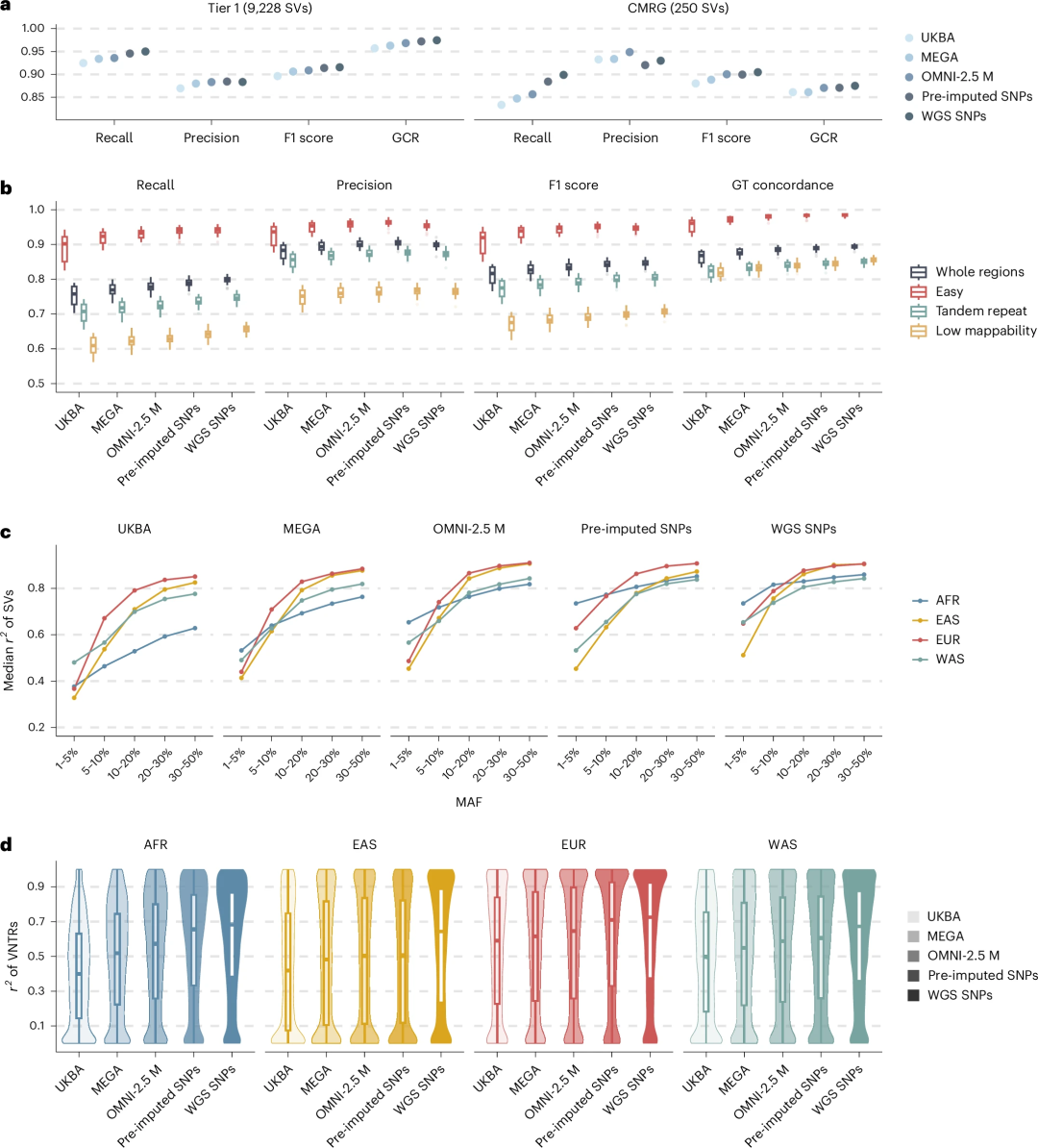
图2.SV插补流程的性能评估。
随后,在UKB的456,643名欧洲血统个体中,研究团队对2,624个性状进行了SV- GWAS。结果共鉴定出17,335个SV-性状关联关系,涉及4,397个SV和656个性状,超过50%性状相关SV与多个性状存在关联(图3)。这些SV至少贡献了复杂性状常见遗传变异的4.7%。其中,位于4q24、9q34.2和7p22.2基因组位点的3个最具多效性的SV与50余个性状相关,主要涉及脑灰质、血液生化指标及穹窿微结构完整性。与其他SV相比,这些性状相关SV不仅长度显著更长,且更靠近转录起始位点和剪接位点。与大规模短读长测序研究相比,该SV关联结果凸显了基于高覆盖HiFi技术的检测panel在解析复杂基因组区域方面的互补优势。
研究团队进一步探究了特定性状相关SV的功能意义,评估了这些变异在多种组织和细胞类型中不同功能基因组类别中的富集情况。与其他类型的SV相比,在大多数细胞类型中,性状相关SV在转录活跃区域及功能性调控元件中显著富集(图3d-e),在组成型异染色质及转录活性稳定受抑制的区域明显减少。同时,这些SV可能通过影响染色质接触或蛋白质与DNA/RNA38的结合来调控性状(图3f-g)。此外,将SV插补分析流程应用于Genotype-Tissue Expression(GTEx;v8)基因型数据进行SV-eQTL分析显示,38.8%的性状相关SV在至少一种GTEx组织中属于SV-eQTLs。研究揭示,CD14+单核细胞中,位于H3K27ac和H3K4me1修饰区域内的一个基因间缺失与哮喘的关联程度,高于该基因位点的所有遗传变异。
研究人员还进行了VNTR与UKB性状的全基因组关联分析。通过VNTR插补流程处理UKB欧洲人群数据,共插补出16,448个VNTR位点,其中84.1%存在于参考panel的欧洲人群基因组组装版本中。在349,662名无血缘关系个体中,研究人员将VNTR与2,624项UKB性状进行关联分析,共鉴定出7,295个VNTR关联位点,涉及1,861个VNTR位点和831个性状,其中1,040个VNTR与至少两种性状相关,1,017个是VNTR -eQTLs。
与一项短读长测序VNTR研究对比显示,基于HiFi的检测panel在稳健的VNTR发现方面具有独特优势,能够补充大型短读长panel所捕获的广泛等位基因多样性。
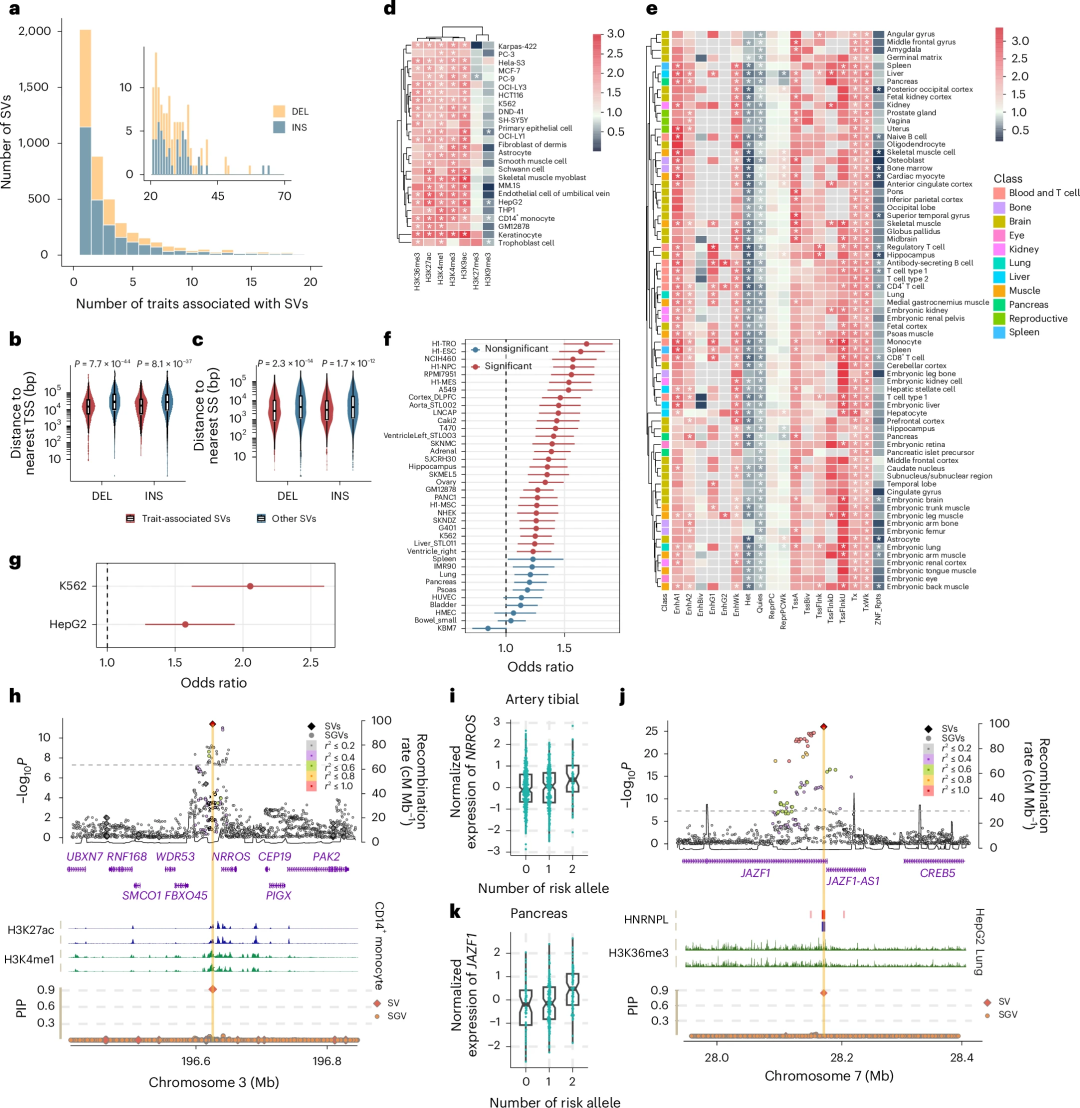
图3.与特定性状相关的SV。
研究团队基于组装数据构建了参考panel,并开发了在线SV插补工具ImputeSV,可为大型队列的复杂基因组区域SV提供精准插补。该研究证明了利用长读长测序基因组组装技术从SNP插补全基因组SV的可行性。基于组装数据的参考panel不仅能通过减少单倍型定相误差来提高插补准确性,还能以高精度获得SV序列信息,同时释放了海量的现有SNP数据集在SV研究中的应用潜力。研究团队公开了参考panel和插补工具ImputeSV,为基于SV的全基因组关联研究开辟了新途径,可应用于基因表达、表观基因组修饰及蛋白质丰度等分子表型分析,也为将SV纳入遗传风险预测模型提供了可能。
ImputeSV公开获取网址:https://yanglab.westlake.edu.cn/ImputeSV
SV- GWAS关联结果:https://yanglab.westlake.edu.cn/data/ukb_sv_gwas
原文信息:
Bai, WY., Liu, S., Duan, Z. et al. Genome-wide associations of structural variants with human traits through imputation from long-read assemblies. Nat Genet (2026). https://doi.org/10.1038/s41588-026-02612-z