首页 > 疾病防控/ 正文
深度解析医学证据,lxfs.net为你支撑决策
1病例资料
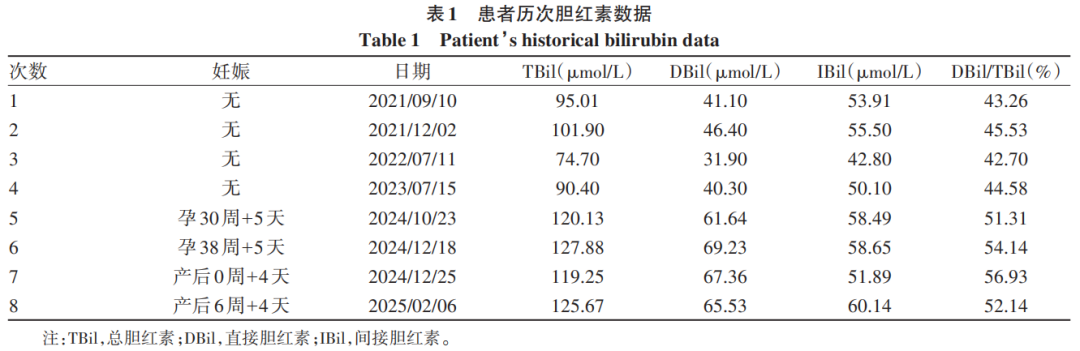
患者女性,30岁,于2025年2月24日因“黄疸30年,脾大1年”在河北医科大学第二医院门诊就诊。患者自出生即出现眼黄、尿黄及皮肤黄,无皮肤瘙痒;劳累、饥饿及饮水少时黄疸加重,妊娠期间胆红素水平较之前升高。患者曾有轻度贫血,已恢复正常,目前无不适主诉。发病以来患者共行8次肝功能检查,最高总胆红素(TBil)为127.88 μmol/L、最高直接胆红素(DBil)为69.22 μmol/L、最高间接胆红素(IBil)为58.65 μmol/L。其中,4次以IBil为主,IBil/TBil比值波动于54.47%~57.30%;4次以DBil升高为主,DBil/TBil比值波动于51.31%~56.93%。既往1年前发现脾大。家族史方面,母亲、双胞胎哥哥均有胆红素升高病史,但无贫血。患者末次月经为2024年3月22日,于2024年12月21日产下1名女性新生儿,新生儿目前无贫血。
门诊查体:全身皮肤、巩膜黄染,浅表淋巴结未触及肿大;双肺呼吸音清晰,未闻及干湿性啰音。腹平坦、柔软,无腹壁静脉曲张,无压痛、反跳痛及肌紧张,未触及包块。肝脾未触及,墨菲征阴性,肾区无叩击痛,无移动性浊音。肠鸣音正常。
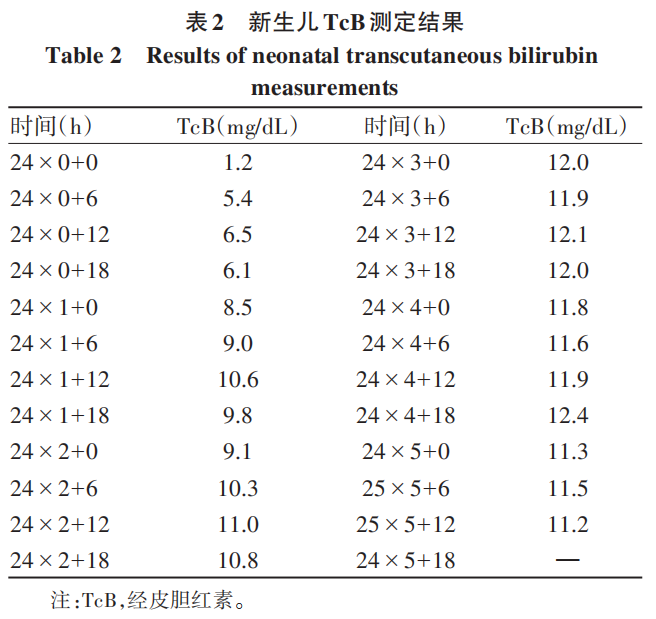
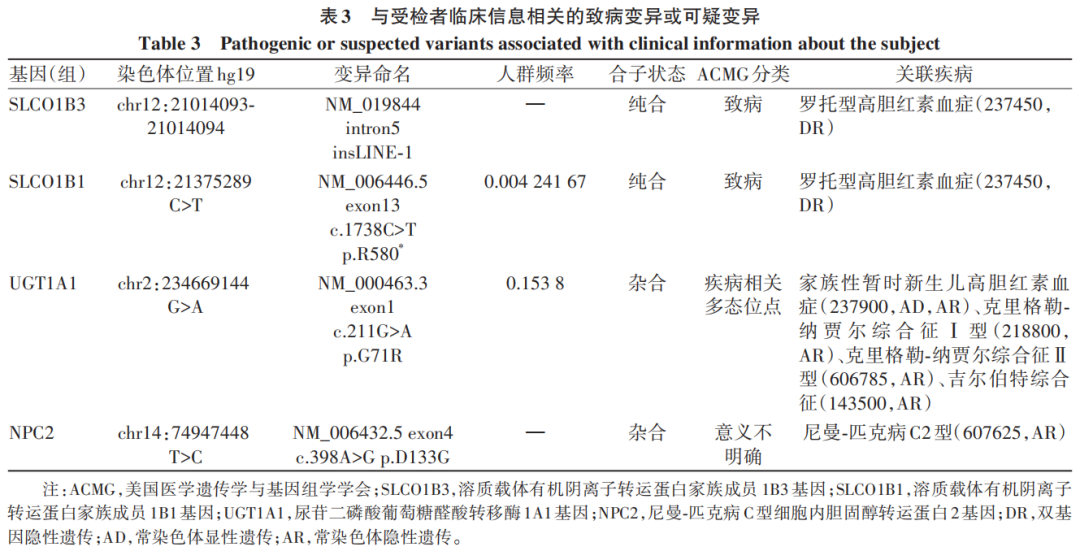
辅助检查:乙型肝炎五项:乙型肝炎病毒表面抗体为229.0 mIU/mL,余四项阴性;丙型肝炎病毒抗体、自身免疫性肝病抗体阴性,血常规、凝血常规、甲状腺功能及转氨酶正常,丙氨酸氨基转移酶为7.5 U/L,天冬氨酸氨基转移酶为13.7 U/L,碱性磷酸酶为133.9 U/L,γ-谷氨酰转移酶为6.7 U/L,总胆汁酸为1.6 μmol/L。患者历次胆红素数据见表1。2024年7月体检发现胆囊息肉,脾稍厚。2024年12月18日腹部超声示脾肋下不大,肋上厚约42.6 mm,长径约139.2 mm,脾内回声均匀,脾门静脉直径约6.9 mm。提示:(1)胆囊前壁强回声(胆固醇息肉或附壁结石),胆囊内少许淤积物;(2)脾大;(3)肝及双肾未见占位性病变。对本例患者产下的足月新生儿每日行经皮胆红素监测(表2),结果均在正常范围。委托天津恩吉思医学检验实验室对患者进行二代测序,通过遗传性肝病基因筛查,发现4个与患者相关的致病变异或可疑变异(表3);并进行一代桑格测序验证,发现染色体chr12:21375289 溶质载体有机阴离子转运蛋白家族成员(SLCO)1B1基因(SLCO1B1)上出现c.1738C>T纯合突变(图1);对SLCO1B3基因突变进行聚合酶链反应产物琼脂糖凝胶电泳验证,SLCO1B3基因5号内含子区域存在大片段插入突变LINE-1(图2)。
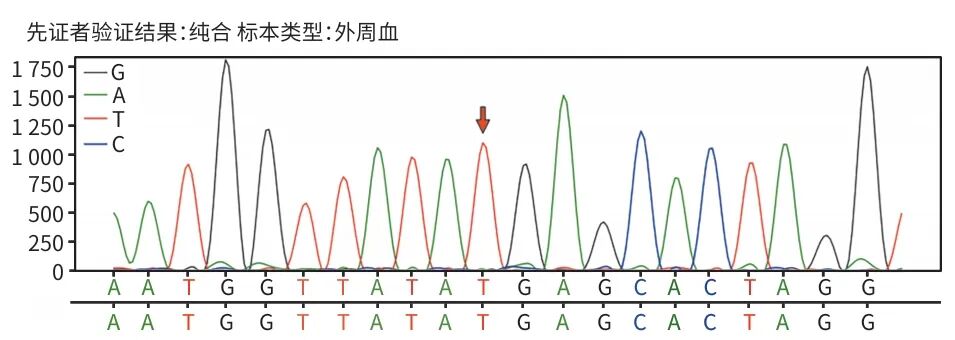
注: 验证位点为SLCO1B1 NM_006446.5:exon13:c.1738C>T:p.R580*,染色体位置为chr12:21375289C>T。红色箭头为突变位点。
图1 桑格测序结果
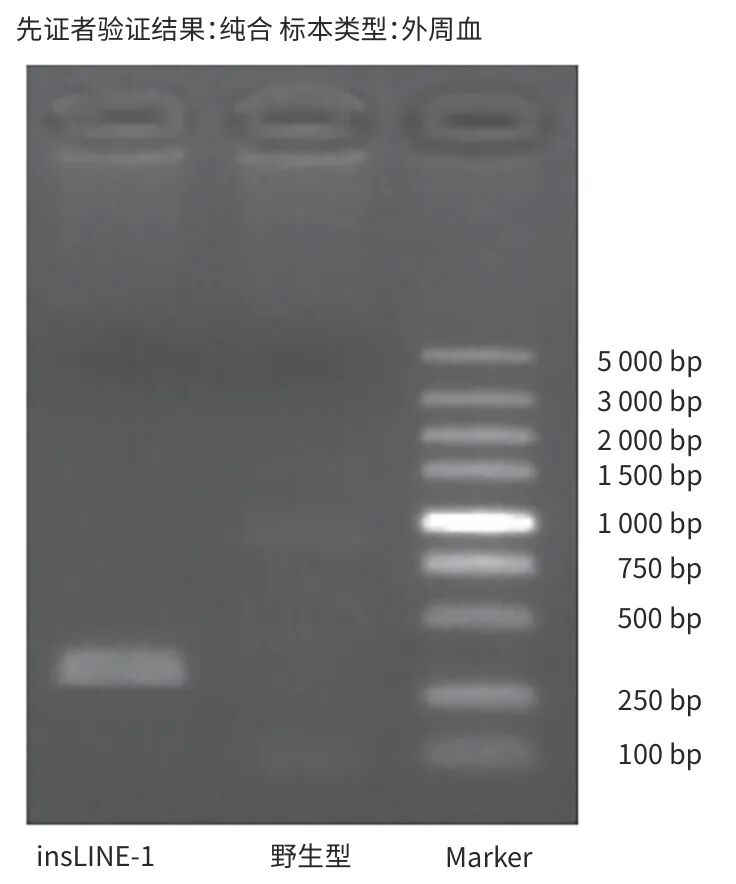
注: SLCO1B3基因5号内含子区域大片段插入突变L1 insertion;染色体位置chr12:21014093-21014094。SLCO1B3,溶质载体有机阴离子转运蛋白家族成员1B3基因。
图2 聚合酶链反应产物琼脂糖凝胶电泳结果
根据患者的临床表现、辅助检查及家族史,综合分析后诊断为罗托综合征。患者自幼出现皮肤、巩膜黄染,无瘙痒,前期以IBil升高为主,妊娠期间胆红素水平有所升高,并转以DBil升高为主;转氨酶、自身免疫性肝病抗体、肝炎检查未见异常,血常规未见异常,无胆道梗阻,可与肝损伤相关的其他肝病相鉴别。基因检测发现SLCO1B1和SLCO1B3双基因纯合突变,进一步支持罗托综合征的诊断结果。此外,患者存在尿苷二磷酸葡萄糖醛酸转移酶1A1基因c.211G>A杂合突变,前期出现以IBil升高为主的混合型高胆红素血症,IBil/TBil比值曾波动于54.47%~57.30%,不符合经典罗托综合征表现,高度怀疑患者部分非胆红素升高由c.211G>A导致酶活性下降引起。患者劳累、饥饿及饮水少时黄疸加重,因吉尔伯特综合征较罗托综合征受应激因素影响波动明显,并且患者伴有单个杂合突变,考虑为吉尔伯特综合征携带者,但暂不考虑诊断可能。尼曼-皮克病C型细胞内胆固醇转运蛋白2基因突变可引起胆固醇和其他脂质在肝脏积累,造成肝细胞损伤、胆汁淤积,间接引起胆红素升高及肝脏肿大。但患者脾稍大、胆囊内少量淤积物仅为近1年出现,且此患者为杂合突变,故暂不考虑尼曼-匹克病。尼曼-匹克病发病较早且伴有神经精神症状,后续应注意监测患者神经精神变化。
2讨论
罗托综合征是一种极为罕见的遗传性疾病,其发病机制是位于12号染色体的双等位基因SLCO1B1、SLCO1B3同时突变,导致位于肝细胞窦膜上的有机阴离子转运多肽(OATP)1B1和OATP1B3功能缺陷,进而引起肝脏摄取和储存缺陷。因此,罗托综合征以粪卟啉尿症和肝脏对多种诊断化合物的摄取显著减少为特征。
罗托综合征的发病机制可以解释其诊断特征:(1)阴离子诊断染料(如溴磺酞钠)血浆清除率降低,临床上表现为溴磺酞排泄试验肝摄取染料延迟,45 min潴留率可高达50%~60%,且在90 min后未出现双相峰。但由于此项检查特异性低且可能产生严重副作用,已不再用于临床试验。(2)阴离子胆汁闪烁显像放射性示踪剂(如99mTc-HIDA)对肝脏的可视化减少,表现为肝脏、胆管和胆囊均不显影,肝脏摄取缓慢,心脏血池持续可视化,肾脏排泄突出。(3)尿粪卟啉(粪卟啉Ⅰ和Ⅲ可能是OATP1B的底物)排泄增加,24 h尿粪卟啉水平上升2~5倍,其中Ⅰ型粪卟啉占总数的65%以上。
近年来,基因检测技术的发展可为罗托综合征早期诊断提供依据,早期行基因检测可与其他肝胆疾病进行有效区分,以明确诊断,从而帮助患者减轻焦虑,避免不必要的检查及治疗。研究显示,只有SLCO1B1和SLCO1B3两个等位基因完全缺乏才会导致罗托综合征,概率仅为1/106,单个基因缺乏不会引起黄疸。检索2000年1月—2025年1月发表的中英文文献,明确诊断为罗托综合征并行基因检测者共19例,其中男性12例,发病年龄为出生后3天~13岁。对患者病例资料进行回顾性分析,发现患者均有皮肤、巩膜黄染(19/19),其中2例患者合并慢性乙型肝炎病毒感染。此外,肝功能显示84.2%(16/19)的患者DBil/TBil为53.1%~93.0%,3例患者DBil/TBil低于50%,分别为48.4%、47.9%和19.3%。84.2%(16/19)的患者提及曾行腹部超声检查,其中68.8%(11/16)的患者腹部超声未见异常,另5例分别表现为胆囊壁稍毛糙、胆囊壁毛糙胆囊结石、脂肪肝伴胆囊隆起样病变、肝脏回声轻度增加和左肾肾盂分离。21.1%(4/19)的患者行肝脏活组织检查,其中2例合并慢性乙型肝炎,10.5%(2/19)的患者行肝胆核素扫描均表现为肝脏、胆管和胆囊均不显影;2例患者行尿粪卟啉检测,1例表现为尿粪卟啉Ⅰ、Ⅲ升高,1例表现为尿粪总卟啉水平正常,异构体Ⅰ占总数的86%,为主导地位,无患者行溴磺酞排泄试验。既往病例报道中,患者基因检测的数据包括:63.2%(12/19)的患者如本例出现SLCO1B1中c.1738C>T和SLCO1B3中5号内含子区域大片段插入,1例出现SLCO1B1中c.1738C>T和SLCO1B3中外显子4缺失,1例SLCO1B1外显子和SLCO1B3外显子纯合缺失,1例SLCO1B1中的c.757C>T与SLCO1B3中的c.1747+1G>A相连,SLCO1B1中的c.1738C>T和SLCO1B3中的LINE-1插入相连,1例SLCO1B1中的c.757C>T、c.1622A>C杂合突变,SLCO1B3中c.1747+1G>A、LINE-1插入杂合突变,1例SLCO1B1中的c.757C>T和SLCO1B3中的c.1747+1G>A相连,1例SLCO1B1中的c.1738C>T、c.1622A>C纯合突变,仅有1例患者二代测序未检测到涉及罗托综合征的SLCO1B1和SLCO1B3基因序列,最终通过肝胆核素扫描结果诊断罗托综合征。综合上述分析,本病例患者与罗托综合征患者高度一致,可诊断为罗托综合征。此患者基因检测存在c.211G>A杂合突变,有研究证明,p.G71R突变通常以纯合状态才能引起吉尔伯特综合征;但也有研究报道,我国吉尔伯特综合征患者中其发生率接近30%,且以杂合为主,杂合的p.Gly71Arg变体可以将尿苷二磷酸葡萄糖醛酸转移酶活性降低至30%~60%,可以解释患者早期以IBil水平升高为主。由于吉尔伯特综合征和罗托综合征均为良性疾病,怀疑遗传性黄疸并不是肝脏活组织检查的指征,对强烈怀疑杜宾-约翰逊综合征/罗托综合征的患者,通常不需要进一步的诊断性评估。该患者罗托综合征诊断已明确,为避免对患者造成不必要的创伤,未再进一步行经皮肝脏活组织检查以排除吉尔伯特综合征。
微小的基因变异可能导致药物毒性的易感性增加,通过基因检测明确发病机制,可避免相关药物引起不良反应。结合本例患者的情况,根据其突变位点及生理机制,对妊娠/哺乳期女性用药和患者日常用药的注意事项及需警惕的具体药物剂量调整原则进行相应的总结,以期为临床治疗提供相应依据。(1)明确OATP1B1/OATP1B3抑制剂及底物的分类。OATP1B1和OATP1B3底物具有高度重叠性,参与大量药物代谢过程,可增加大剂量环孢菌素A、阿利泊韦、抗癌剂、甲氨蝶呤等抗病毒药物以及他汀类药物的血浆和组织浓度,同时SLCO1B1变体是羟基甲基戊二酸酰辅酶A还原酶抑制剂不良反应(如肌病或横纹肌溶解症)的危险因素,特别SLCO1B1 5219 T>C基因变异是辛伐他汀相关肌病风险最重要的预测因子。此外,SLCO1B3基因内含子LINE-1插入可导致个体肝脏对吲哚菁绿的摄取严重受损,引起抗癌药物的耐药性。多种常用药物可作为其抑制剂或底物,OATP1B底物包括普伐他汀、阿托伐他汀、辛伐他汀、瑞舒伐他汀、氟伐他汀、缬沙坦、利福平、格列本脲、依折麦布、甲氨蝶呤、紫杉醇、伊马替尼、地高辛和多西他赛,伊立替康代谢产物SN-38,顺铂;抑制剂包括利福平、环孢素、克拉霉素、利托那伟、吉非罗齐和熊去氧胆酸,因此,明确OATP1B1/OATP1B3抑制剂及底物的分类对临床用药具有重要意义。(2)避免使用导致胆红素从白蛋白中置换的药物,例如异丙酚、磺胺类药物、头孢曲松、氨苄西林、水杨酸盐和呋塞米,或被认为具有肝毒性的药物。(3)罗托综合征患者应用部分OATP1B抑制剂及底物时可根据患者情况考虑更低的药物剂量,特别是关注治疗指数窄可能会导致显著的临床结局的高风险药物,如利福平联合依折麦布给药会导致体内OATP1B1的额外抑制。Bouchghoul等研究发现,在接受格列本脲治疗的妊娠糖尿病患者中,OATP1B3遗传变异的女性与较高的低血糖风险和较低的格列本脲剂量相关。然而,部分药物仅增加结合胆红素的血浆浓度,而未见有其他肝损伤证据。因此,在应用此类药物时可据实际情况综合适时调整剂量。(4)肾功能受损患者的药物非肾清除率进一步降低,药物用量需更为慎重。Sun等研究发现,尿毒症毒素的积累可以抑制有机阴离子转运蛋白;此外,在慢性肾功能衰竭大鼠的肝脏中,观察到OATP1B蛋白表达降低了35%。目前暂无文献对OATP1B抑制剂及底物的具体用量作出明确的指导,在多药方案中应考虑抑制剂/底物的重叠作用,情况也更为复杂,已有监管指导文件建议评估药物对OATP1B的抑制潜力。共轭卟啉Ⅰ被确立为OATP1B介导相互作用的一级生物标志物,其他内源性底物如共轭卟啉Ⅲ等具有相似性。未来可通过数据建模明确罗托综合征患者与抑制剂及底物的低、中、强相关性,从而精准预测用药剂量。
罗托综合征患者预后良好,当出现并发疾病、口服避孕药和怀孕时,可能会引发黄疸,但对母体或胎儿的预后无影响,一般无需给予特殊治疗。本例新生儿出生后,每日行经皮胆红素监测,结果均在正常范围。目前未检索到罗托综合征患者妊娠相关的文献,可暂时参照遗传性高胆红素血症克里格勒-纳贾尔综合征患者的妊娠管理建议,并对此进行相应调整,建议患者:(1)怀孕前讨论潜在问题和遗传咨询;(2)避免使用肝毒性药物,对于OATP1B1/3抑制剂及底物相关药物的用量可据患者情况适量降低;(3)监测新生儿胆红素值;(4)产后避孕咨询时,需考虑激素避孕药经肝脏的代谢消除,会引起高胆红素血症恶化的风险。
罗托综合征是罕见的胆红素代谢障碍性疾病,随着基因检测技术的飞速发展,通过基因检测明确罗托综合征患者基因层面的变异,可以进一步明确其发病机制,及早发现并干预,帮助患者进行产前预防和给予用药指导。本例报告提示临床医生应加深对基因检测等新式手段的认识,对疑似罗托综合征患者尽早行基因检测以明确诊断,同时重视为患者提供个体化治疗方案,达到精准医疗的目标。
全文下载
https://www.lcgdbzz.org/cn/article/doi/10.12449/JCp60321

小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP
