首页 > 疾病防控/ 正文
深度解析医学证据,lxfs.net为你支撑决策
本研究旨在筛查中国疑似视网膜色素变性(RP)患者的致病性变异。本研究纳入了一个包含75名临床诊断为RP的无亲缘关系中国患者及其可获得的家庭成员组成的队列。提取所有受试者的基因组DNA,并应用全外显子组测序(WES)。筛选候选变异,并进行minigene检测以评估新剪接变异的致病性。
总体而言,诊断率为44%(33/75),并发现了16个此前未报道的新变异。在遗传学确诊的33例病例中,31名患者被确定为携带RP的致病性变异,2名患者携带与其他视网膜疾病相关的致病性变异。USH2A、CYP4V2和RPGR是最常见的致病基因,约占遗传学确诊病例的一半。此外,minigene检测验证了新剪接变异是有害的。此外,9名患者在6个常染色体隐性遗传模式基因中携带单个有害杂合变异,且未检测到相应的拷贝数变异(CNVs)。本研究的发现揭示了中国RP的基因图谱,并为临床医生提供了指导。
-
全外显子组测序是视网膜色素变性诊断的有效临床辅助工具;
-
在中国视网膜色素变性患者中,热点基因为USH2A、CYP4V2和RPGR;
-
检测到16个此前未报道的新变异;
-
Minigene检测验证了两个新剪接变异可能破坏了RNA剪接。
研究背景
视网膜色素变性(RP,OMIM #268000)具有广泛的临床和基因异质性,是由感光细胞和色素上皮细胞进行性营养不良引起的一组遗传性眼病(Campochiaro and Mir, 2018)。RP的发病率具有很大的地域和种族差异,全球患病率估计为1/4000(Hartong et al., 2006),在中国患病率约为1/3000(Hu, 1987)。大多数RP患者以夜盲症为最早症状,而少数因视力下降就诊,通常在青春期发病且进展缓慢(Berson, 1993;Ayuso and Millan, 2010;Oishi et al., 2014)。最初可表现为视野缺损和典型的环形暗点。晚期视野呈管状,在中年或老年时完全失明。RP的遗传方式复杂,包括常染色体显性遗传模式(AD,约20%)、常染色体隐性遗传模式(AR,约30%)和X连锁遗传(XL,约10%),偶见双基因模式和线粒体遗传模式。无家族史的病例称为散发病例,散发性RP的比例接近40%(Martin-Merida et al., 2019)。RP的遗传模式也与其严重程度相关。X连锁RP(XLRP)是青少年致盲的常见原因之一,其特征为男性发病早、临床表现严重(Bunker et al., 1984;Cehajic-Kapetanovic et al., 2020)。
根据是否伴有其他症状,RP分为非综合征型视网膜色素变性(NSRP)和综合征型视网膜色素变性(SRP)。前者仅限于视觉异常,而后者涉及其他遗传综合征,如Usher综合征(Huang et al., 2017;Kimchi et al., 2018;Verbakel et al., 2018)。NSRP是最常见的遗传性视网膜变性,约占RP的70%(Bunker et al., 1984;Kimchi et al., 2018)。迄今为止,研究已报道了90多个与RP相关的致病基因(RetNet, https://sph.uth.edu/retnet/),这些基因对应特定的疾病亚型,可能具有特定的发病年龄、视力损害和疾病进展(Verbakel et al., 2018)。然而,由于RP与其他遗传性视网膜营养不良疾病在临床表现上的相似性,分子基因学检测对于明确诊断是必要的。
二代测序技术已在分子诊断中得到广泛应用,尤其是全外显子组测序(WES)。WES已被证明是诊断遗传性疾病的有力工具,并在遗传性疾病的诊断中发挥着不可或缺的作用(Yang et al., 2013;Carss et al., 2017)。本研究对75名疑似RP患者进行了WES,以寻找疾病的分子病因。
研究结果
人口统计学:
本研究共纳入75名无亲缘关系的RP患者(包括37名男性和38名女性)及其可获得的家庭成员。患者年龄范围为4至68岁,平均年龄为44.6±11.2岁。所有初步临床诊断为RP的患者均有视力下降和夜盲的主诉。本研究中大多数RP病例为散发性,约73.3%无明显家族史,发病年龄分布在不同的阶段(表1)。
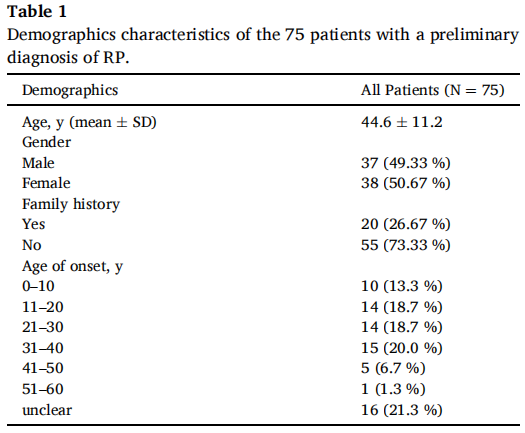
表1
全外显子组测序的基因学发现:
致病性变异筛选流程如图1所示。通过WES识别出每位患者大约13,000个单核苷酸变异或小片段插入/缺失。排除良性变异后,共发现146个与遗传性视网膜疾病相关的突变,包括32个致病性变异、46个可能致病性变异和68个意义不明确的变异。总共有33例病例中的43个不同变异符合潜在致病性变异的条件,总体诊断率为44%(33/75),其中在31名患者中识别出RP的致病基因,在2名患者中识别出其他视网膜疾病的致病基因(图2)。这些变异位于16个基因中:USH2A、CYP4V2、RPGR、EYS、ABCA4、PDE6B、PROM1、IMPG2、IFIT40、RP1L1、PRPH2、HK1、PRPF8、CRX、RPRGIP1和CHM,包含16个新突变(图3)。
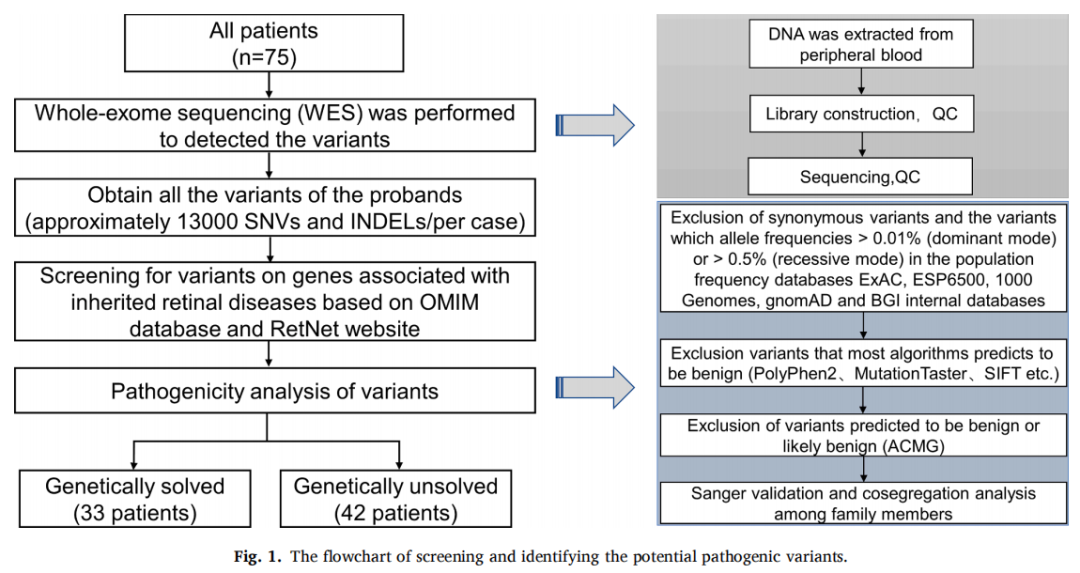
图1
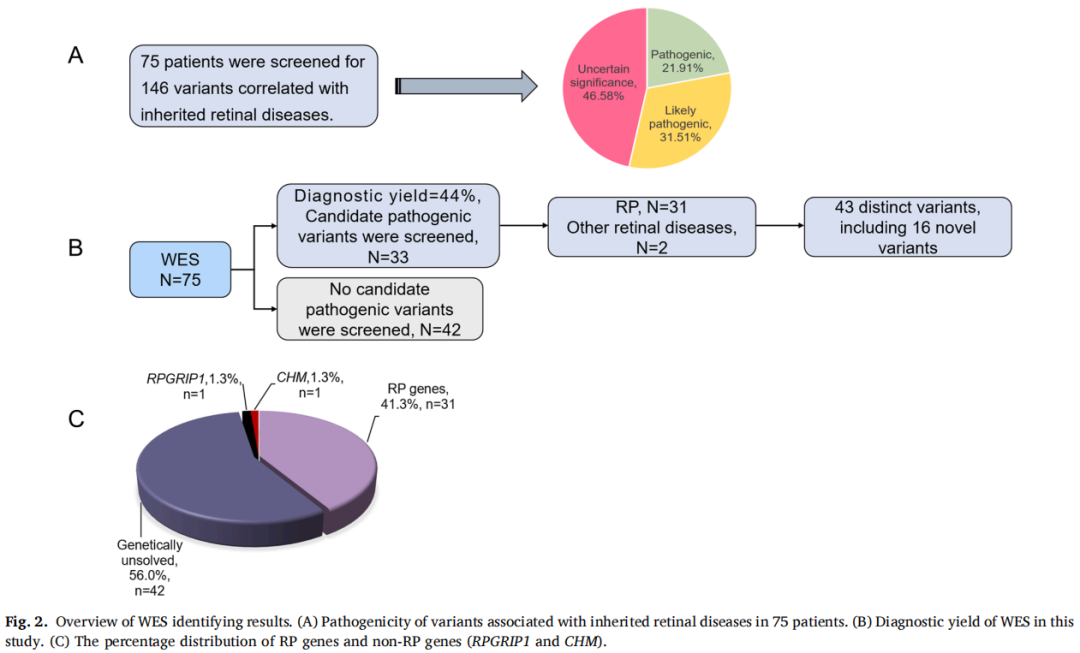
图2
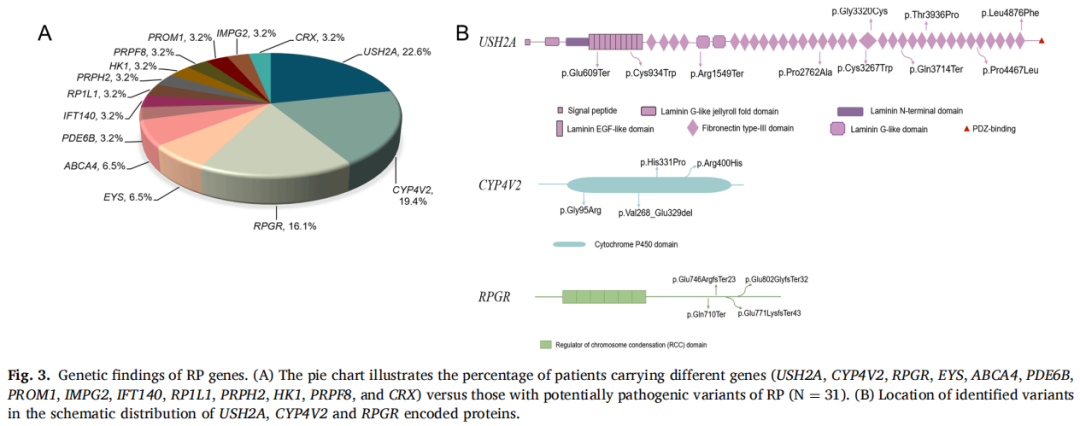
图3
RP基因的基因学发现:
31名患者被确定为在14个已知RP基因中携带致病性突变。就频率而言,排名前三的RP基因为USH2A、CYP4V2和RPGR,占本研究基因学确诊病例的一半以上(图3A)。在三个主要基因上检测到的变异分别表示在其蛋白质结构域上(图3B)。
能够解释RP的变异如表2所示,新识别出15个不同变异。31例病例中有18名患者在ARRP基因中携带杂合致病性变异,占大多数,而仅有4例为纯合病例,包括USH2A、CYP4V2、PDE6B和IMPG2基因各一例。同时,在PRPH2、HK1、PRPF8和CRX基因中检测到4个不同变异,涉及常染色体显性遗传模式,仅在5例病例中检测到涉及XLPR的RPGR变异。在RP基因的新变异中,有5个错义突变、2个无义突变、2个剪接突变和6个移码突变。三个新突变发生在ADRP基因中,两个发生在XLPR基因中。其余10个新变异发生在ARRP基因中,包括USH2A、EYS、PROM1、IMPG2、IFIT40和RP1L1。
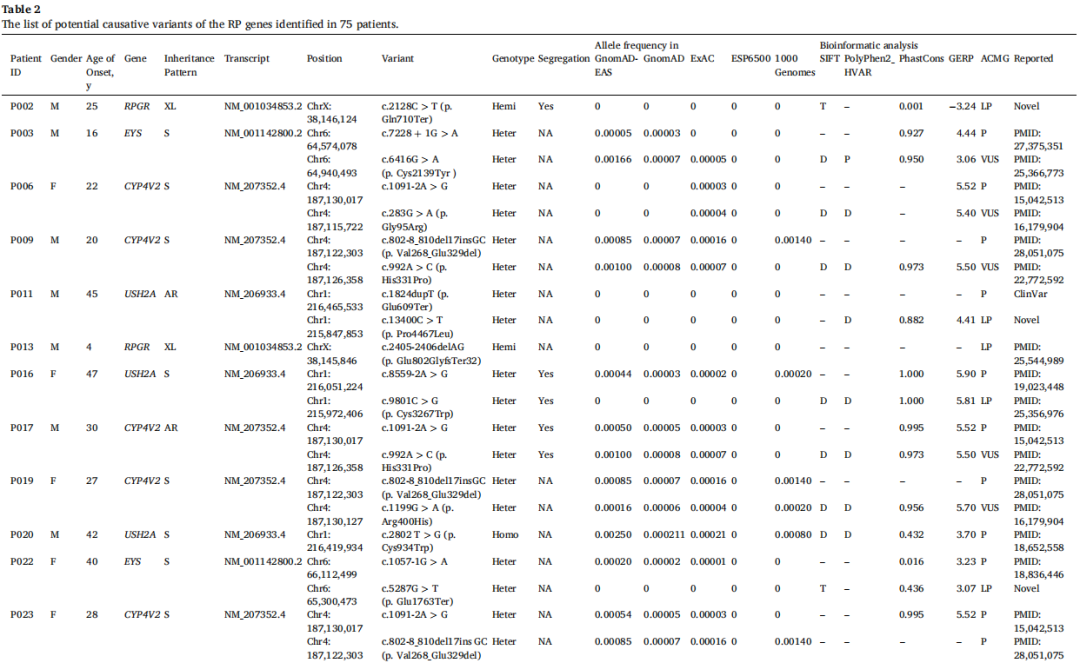

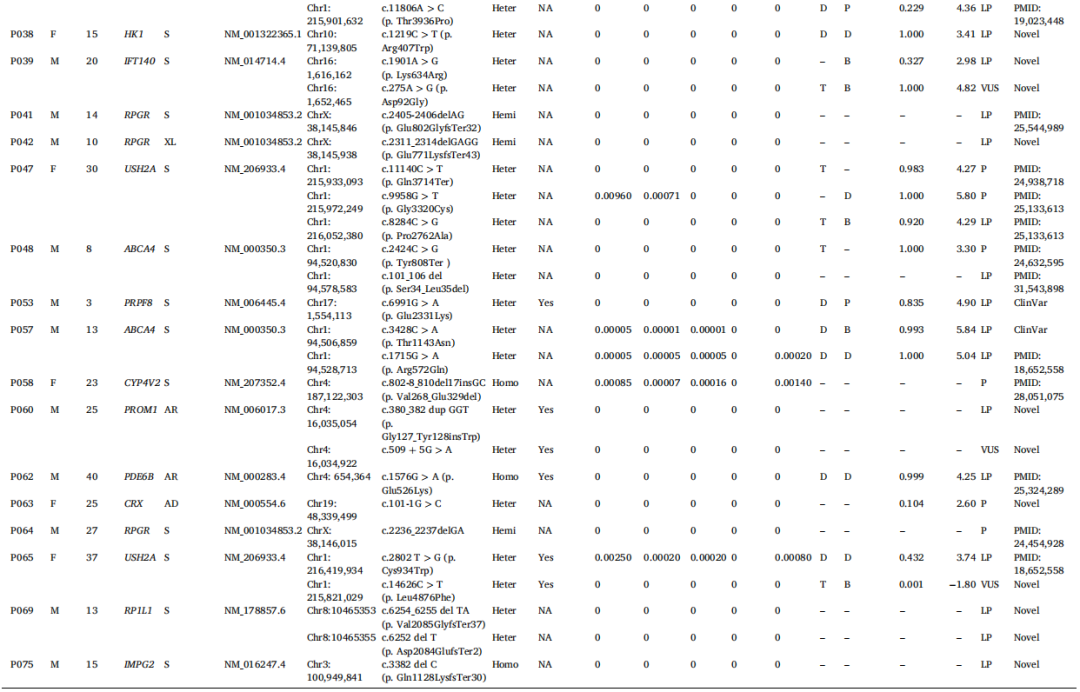
表2
此外,在9名患者中仅检测到与ARRP相关的单核苷酸变异(SNV)。虽然CNV结果表明每位患者携带约30个CNV,但均不位于所识别SNV的同一基因上,且在这些患者中未发现表型相关的CNV(表3)。该结果排除了由CNV和单SNV组合引起的单倍体不足导致致病性的可能性。因此,尽管根据ACMG指南这些SNV被认为是致病性或可能致病性的,但在本研究中它们未被归类为致病性突变。
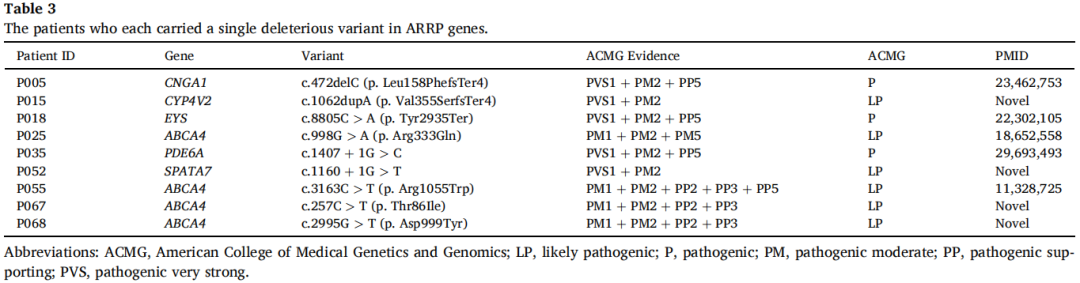
表3
其他遗传性视网膜疾病相关基因中的致病性突变:
此外,在与可能引起其他常染色体隐性或X连锁显性视网膜疾病相关的两个基因中检测到三个突变,包括一个新突变和两个已知突变。患者P014在RPRGIP1中携带复合杂合突变,患者P066在CHM中携带致病性变异c.1244+1G>A,该变异与无脉络膜症相关(表4)。
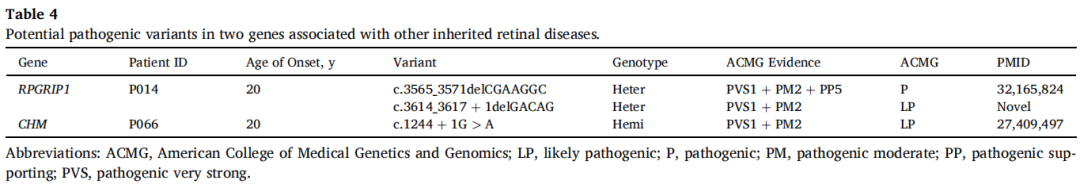
表4
RPRGIP1已被报道与常染色体隐性遗传视网膜营养不良疾病相关,包括Leber先天性黑矇和视锥-视杆营养不良。CHM是X连锁遗传无脉络膜症中唯一报道的致病基因。在一定程度上,上述疾病与RP在临床特征上有一定相似性,需要通过基因检测进行鉴别诊断。患者P014和P066均有视力下降、夜盲、无眼球震颤,且在出生后二十年开始出现症状。考虑基因检测结果,前者被重新分类为视锥-视杆营养不良,后者被重新评估为无脉络膜症。
新剪接变异的Minigene功能检测和新错义及插入变异的蛋白质结构预测:
进行Minigene检测以验证新剪接变异的有害性,结果表明PROM1中的变异c.509+5G>A和CRX中的变异c.101-1G>C对正常的RNA剪接产生不利影响。PROM1中的变异c.509+5G>A和CRX中的c.101-1G>C导致异常剪接,分别触发外显子4和外显子3的缺失,如图4所示。
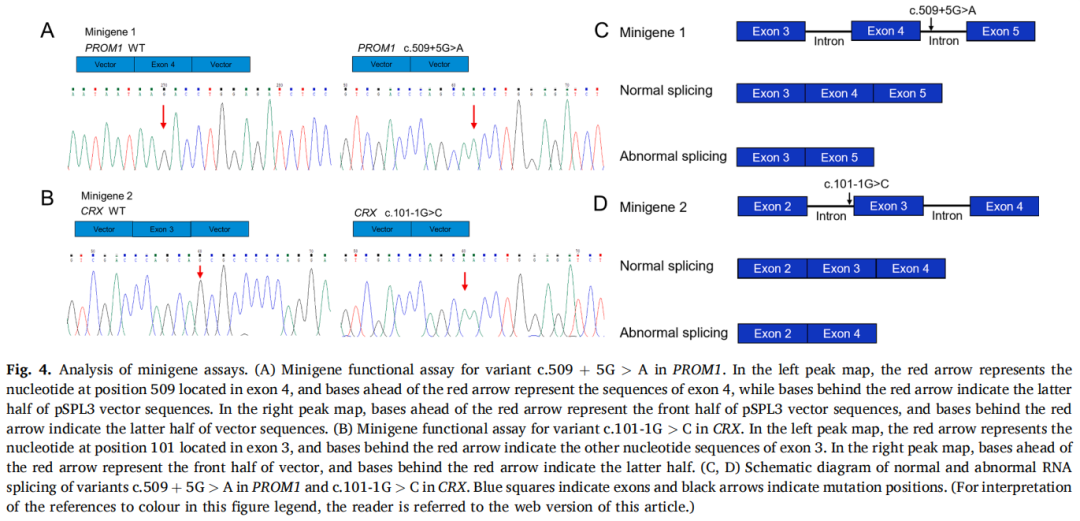
图4
由新错义和插入变异引起的蛋白质二级结构改变通过ANTHEPOT软件(Version 6.93)预测,如图5所示。
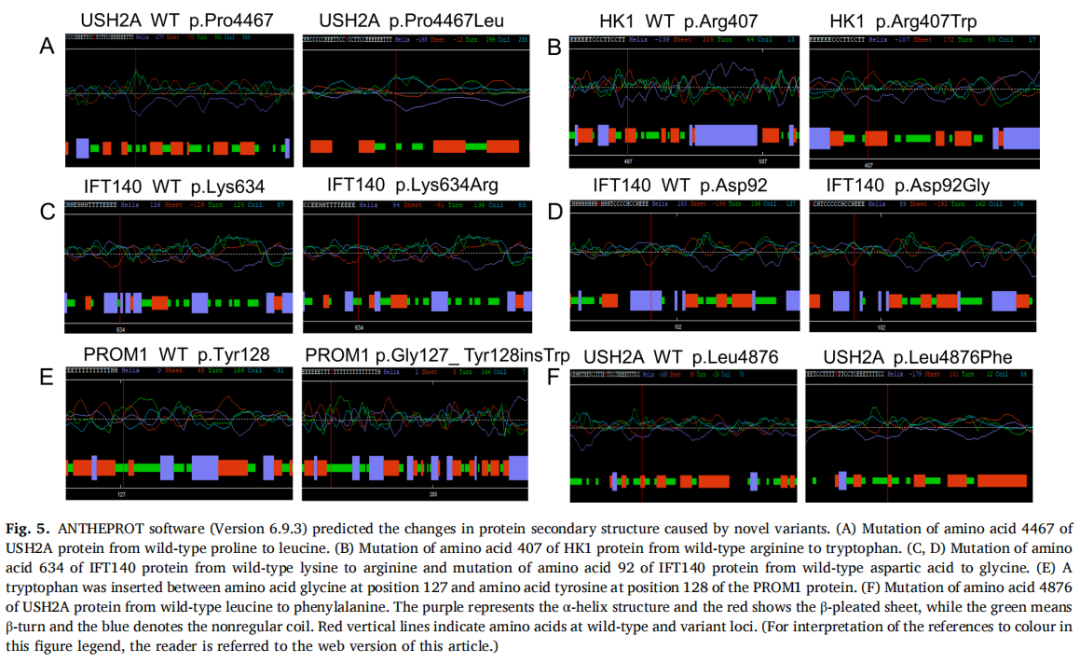
图5
讨 论
RP是最常见的遗传性视网膜营养不良(IRD)形式,其发病年龄、严重程度和进展与遗传和遗传模式相关,发病机制复杂且不明。据报道,IRD与约300个基因相关,其中90多个基因为RP致病基因(Kim et al., 2021)。仅从临床表现准确区分RP和其他视网膜营养不良疾病具有挑战性。基因检测可有助于鉴别诊断。
在本队列研究中,对75名疑似RP的无亲缘关系患者进行了WES。总体而言,诊断率为44%,接近先前文献报道的平均水平(Neveling et al., 2012;Xu et al., 2014)。共有31名患者被确定为在已知RP基因中携带潜在致病性变异,包括15个新变异。约一半的基因学确诊病例归因于3个基因的变异:USH2A、CYP4V2和RPGR。本研究中检测到的最常见的已知RP致病基因是USH2A,这与先前在中国人群中的报道一致(Huang et al., 2017;Gao et al., 2019;Dan et al., 2020)。然而,EYS是日本RP患者中最常检测到的基因(Oishi et al., 2014),而FAM161A在以色列-犹太人群中的突变频率最高(Beryozkin et al., 2020)。这表明RP致病基因的患病率在中国人和其他种族中存在差异,而USH2A、CYP4V2和RPGR可能是中国的热点基因。
据报道,在北美和西班牙,USH2A变异已被确定为7-23%的非综合征型RP的致病基因(Zhu et al., 2020),并且它也是中国RP的主要原因。USH2A基因位于人类常染色体1q41上,由72个外显子组成,编码一种存在于基底膜中的蛋白质,在视网膜和前庭发育中发挥重要作用(Santana et al., 2019)。USH2A基因的突变谱非常异质,涉及1500多个突变,其中超过690个突变被认为是致病的(Toualbi et al., 2020)。在本研究中,7名患者中有6名携带USH2A杂合变异,其余1名为纯合。突变c.8559-2A>G、c.2802 T>G(p.Cys934Trp)、c.9958G>T(p.Gly3320Cys)和c.8284C>G(p.Pro2762Ala)由不止一名患者携带,可能是中国人的热点突变。迄今为止,USH2A基因型与中国患者表型之间的关系尚未被充分描述。在本研究中,携带USH2A基因致病性突变的患者发病年龄各异。此外,患者P047表现出高频下降的听力损失症状,而低频无明显异常,被怀疑为综合征型RP,与USH2A突变可能引起的综合征表型一致。
本研究中频率仅次于USH2A的RP基因为CYP4V2。CYP4V2基因负责编码细胞色素p-450蛋白家族的成员,该家族在脂肪酸代谢中发挥作用,并在视网膜色素上皮细胞中高表达(Nakano et al., 2012)。由CYP4V2突变引起的Bietti结晶样视网膜病变(BCD)是RP的一种特殊表现,BCD患者在东亚地区(尤其是中国和日本)相对常见(Hu, 1983;Lin et al., 2005)。在CYP4V2基因中识别出5个不同变异:c.1091-2A>G、c.283G>A(p.Gly95Arg)、c.802-8_810del17insGC(p.Val268_Glu329del)、c.992A>C(p.His331Pro)、c.1199G>A(p.Arg400His)。变异c.802-8_810del17insGC在4名无亲缘关系患者中检测到,该变异在东亚BCD患者中高频出现(Jiao et al., 2017;Zhang et al., 2018)。此外,c.992A>C(p.His331Pro)在2名患者中发现,c.1091-2A>G分别在3名无亲缘关系病例中识别。值得注意的是,这些变异通常仅在东亚患者中报道(Zhang et al., 2018),这暗示突变谱具有种族特异性。
第三常见的变异基因是RPGR,它引起XLRP,其特征为早发和快速视力丧失的严重表型(Cehajic-Kapetanovic et al., 2020;Salvetti et al., 2021)。检测到5名患者携带RPGR变异,其中两人各携带一个新突变c.2128C>T(p.Gln710Ter)或c.2311_2314delGAGG(p.Glu771LysfsTer43)。本研究中所有携带RPGR突变的患者均为男性,且发病年龄早于携带USH2A基因突变的患者,其他三名患者在罹患RP时年龄小于15岁。这些变异均位于开放阅读框15(ORF15)外显子上,导致RPGR蛋白过早截短。该结果与其他研究一致,报道富含嘌呤的ORF15区域是突变热点(Yang et al., 2014)。在英国人群调查中,XLRP患者ORF15区域的突变率接近80%(Vervoort et al., 2000;Bader et al., 2003)。
同时,在9名患者中检测到不同ARRP基因中的单个有害SNV,主要富集在ABCA4基因中,这些不能归类为这些个体的致病原因。这些结果可能归因于WES不可避免的局限性,即排除非编码区、深内含子区和CNV的检测。据报道,CNV增加了隐性遗传病的发病率,并在遗传性视网膜变性中贡献了9%的致病性(Zampaglione et al., 2020),因此进一步分析了这些个体是否携带单个有害SNV和致病性CNV。不幸的是,未获得与单SNV基因相对应的CNV,在这9名患者中也未追踪到与疾病表型相关的CNV。
此外,在2名疑似RP患者中检测到位于RPGRIP1和CHM基因上的致病性变异,这些基因与其他视网膜疾病相关,更新了之前RP的表型诊断,并说明了基因检测在疾病诊断中的重要性。此外,通过minigene检测验证了位于PROM1和CRX基因上的新识别剪接变异,证明这些变异可能破坏了RNA剪接。
尽管如此,本队列研究仍存在一定的缺点,需要进一步解决。首先,本研究中未确诊的病例未能阐明疾病的分子病因。其次,WES存在一些不可否认的局限性,例如RPGR的ORF15区域覆盖不足(Huang et al., 2015)。此外,患者的发病年龄为自我报告,可能存在主观性和偏倚。接下来,由于若干客观因素,缺乏对候选致病性变异的分离分析。值得注意的是,诊断率在实际意义上并不等同于真实诊断率。例如,在本研究中,在患者P074中识别出ARRP基因DHX38的两个变异,但它们均遗传自其母亲,不能视为RP的致病基因。因此,在父母或家庭成员中的验证在分子诊断中至关重要。最后,本队列的样本量相对较小也是本研究的局限性之一,使研究者无法获得更多关于基因型-表型关联的信息。
总之,本研究表明中国RP患者中频率较高的致病基因为USH2A、CYP4V2和RPGR。同时,共检测到16个新变异,并在体外验证了2个新识别的剪接变异。本研究的发现不仅为中国RP的基因谱提供了可靠的见解,而且还阐明了分子诊断的价值及其在未来诊断、遗传咨询和个体化治疗中的贡献。
参考文献:
Jin B, Li J, Yang Q, et al. Genetic characteristics of suspected retinitis pigmentosa in a cohort of Chinese patients. Gene. 2023;853:147087. doi:10.1016/j.gene.2022.147087
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP
