首页 > 医疗新闻/ 正文
深度解析医学证据,lxfs.net为你支撑决策
非典型畸胎瘤/横纹肌样瘤(ATRT)是一种局限于神经轴的侵袭性恶性肿瘤。该病主要影响婴幼儿和年幼儿童,在青少年和年轻成人中极为罕见。虽然其中枢神经系统播散较为常见,但神经外转移则异常罕见。本文报告一例 16 岁脑ATRT患者的病例,该患者在初次缓解 8 个月后,出现了累及内脏和骨骼部位的神经外复发。这种不寻常的表现给鉴别复发与第二原发性横纹肌样肿瘤带来了重大诊断挑战。使用多基因 panel进行的基因分析比较发现,原发中枢神经系统肿瘤与复发灶存在相同的突变,证实了它们共同的克隆起源,并排除了第二新发恶性肿瘤的可能性。本病例强调了关注ATRT系统性复发可能性的重要性。据研究人员所知,这是首次报告应用比较基因分析来区分转移性复发与第二原发肿瘤。需要更大规模的研究来验证该方法在不同肿瘤类型中的适用性。
背 景
恶性横纹肌样瘤(MRT)是一种高度侵袭性的肿瘤,主要影响婴幼儿和年幼儿童,但偶见于青少年和成人。它们可发生于中枢神经系统(65%)和多个其他器官(35%),其命名由原发肿瘤的部位决定。当位于中枢神经系统时,被称为非典型畸胎瘤/横纹肌样瘤(ATRT)。非中枢神经系统的横纹肌样瘤可发生于肾脏、肝脏、软组织和其它部位。通常,它们被分为肾脏恶性横纹肌样瘤(RTK)和肾外非中枢神经系统横纹肌样瘤(eMRT)。颅外MRT相较于ATRT预后更好,其假设性解释是,对于非中枢神经系统肿瘤,通过广泛手术切缘和放疗通常更容易实现局部控制。
ATRT和非中枢神经系统MRT的基因组标志是SMARCB1(INI-1)的双等位基因失活,或罕见的SMARCA4双等位基因失活,它们编码SWI/SNF染色质重塑复合体的一个核心亚基。由于在MRT中很少报道有其他基因组改变,且SMARCB1/SMARCA4缺失直接影响表观基因组,因此普遍认为MRT的流行病学和临床病理多样性受甲基化模式的高度影响。基于对颅内和颅外MRT多组学数据的无监督聚类,提出了将其分为五个分子组的分类,反映了主要失调的通路:第1组,"ATRT-MYC样";第2组,"ATRT-TYR样";第3组,"RTK样";第4组,"肾外MRT样";以及第5组,"ATRT-SHH样"。一些作者认为存在第六个分子亚组,即SMARCA4突变的MRT,主要与颅外原发肿瘤相关。分子亚组与原发肿瘤的解剖部位之间存在一定的相关性。因此,第 2 组和第 5 组肿瘤主要是ATRT,很少发生于颅外;第3组和第4组倾向于非中枢神经系统部位;第1组在ATRT和非中枢神经系统MRT中均常见。有罕见的继发性ATRT病例报告,发生于胶质瘤或其他中枢神经系统肿瘤的背景上,其中一些与李法美尼综合征相关。
已有报道ATRT与颅外MRT同时性和异时性共存。例如,10%-15% 的RTK患者在诊断时存在同时性ATRT。横纹肌样瘤易感综合征(RTPS)是一种遗传性癌症易感性疾病,其特征为SMARCB1/SMARCA4杂合性基因变异。1型RTPS与基因中央部分的突变相关,涉及多个外显子缺失或重复以及截断突变,主要与颅内和颅外横纹肌样瘤相关。另一方面,迄今为止,仅有少数确切的ATRT在中枢神经系统外复发的病例被报道。本文描述了一例幕上ATRT病例,经多模式治疗后达到完全缓解,随后出现多个中枢神经系统外的横纹肌样瘤病灶,在鉴别ATRT复发与第二原发性颅外MRT方面构成了诊断挑战。
病 例
患者男,16 岁,因头痛、颈部僵硬和呕吐 1 周就诊。术前脑部MRI显示,右侧额顶叶中心有一个边界清晰、厚壁的囊性病变伴壁结节(图1a, b)。随后患者出现神经功能恶化、反射减弱和血流动力学不稳定,需行急诊右侧额顶叶开颅肿瘤切除术。组织病理学检查确诊为非典型畸胎瘤/横纹肌样瘤(ATRT),WHO 4级,其特征为核INI-1表达缺失(图2a–c)。术后MRI显示有残留病灶(图1c、d)。患者转诊以接受进一步治疗。胸腹CT和全脊柱MRI未见转移征象。脑脊液细胞学检查未见恶性细胞。多学科团队(MDT)委员会初步计划按照Chi等人的方案,先行诱导化疗,随后进行手术切除、放疗和巩固化疗。然而,在开始治疗前,患者临床状况急剧恶化,出现发热和颅内高压征象。初次手术后第 39 天复查脑脊液细胞学结果转为阳性。患者接受了第二次手术,对进展性残留脑肿瘤进行最大安全范围切除。肿瘤似乎已长入之前的开颅间隙,浸润至头皮。硬脑膜增厚并被肿瘤浸润。切除受浸润的硬脑膜后,可见肿瘤。肿瘤呈血管性、灰白色、质韧且软,与脑组织分界不清。肿瘤被尽可能分块切除。MDT重新评估后将疾病分期定为M1期,并修订治疗策略,优先进行初始颅脊髓照射而非前期全身治疗。采用容积旋转调强放疗(VMAT)分三个阶段进行颅脊髓照射,总剂量 59.4 Gy,每次 1.8 Gy:第一阶段为全颅脊髓轴照射 36 Gy,分 20 次;第二阶段为手术床加 1 cm边界照射 18 Gy,分 10 次;第三阶段为肿瘤床外加 3 mm边界照射 5.4 Gy,分 3 次(图3a–d)。放疗后脑部和脊柱MRI显示完全缓解。患者继续接受放疗后的多药联合化疗方案,包括长春新碱、多柔比星、环磷酰胺、放线菌素D和替莫唑胺。
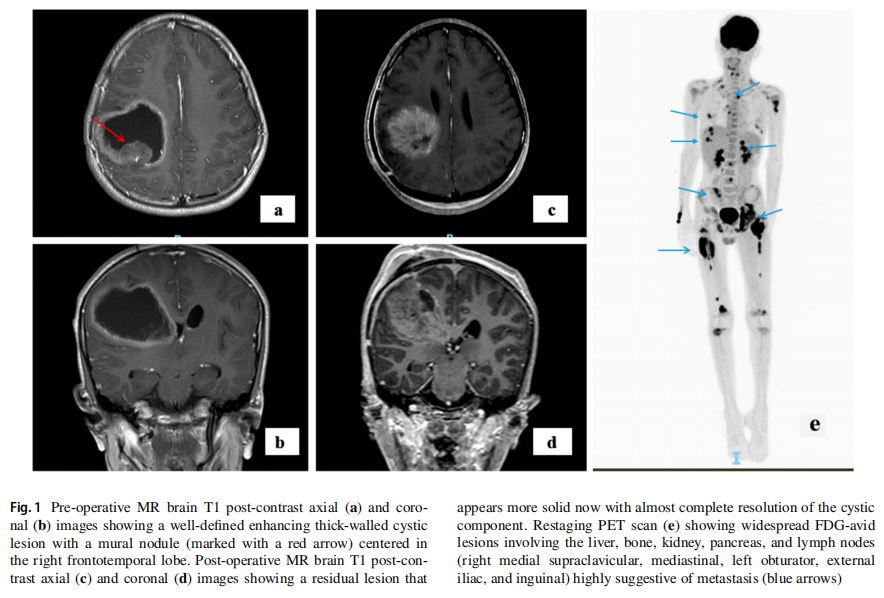
图1 影像学检查结果
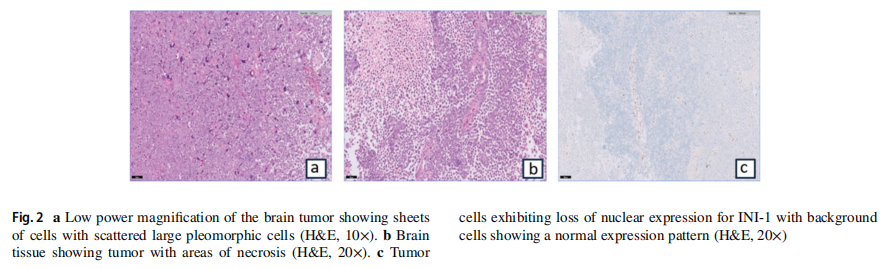
图2 组织学检查结果
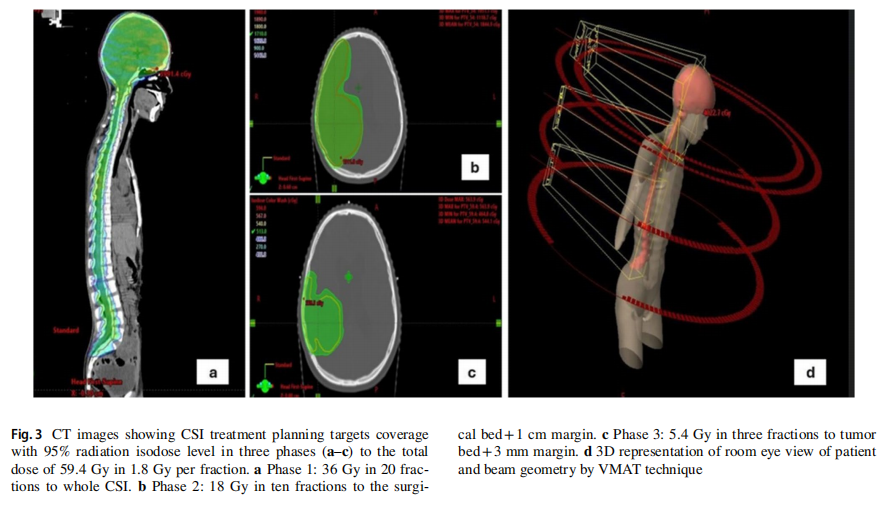
图3 CT图像显示CSI治疗计划靶区在三个阶段(a–c)中以95%辐射等剂量水平覆盖,总剂量为59.4Gy,每次分割剂量为1.8Gy。
患者临床状况逐渐改善,变得无症状且功能完全正常。他按计划接受了多药化疗方案直至第 30 周。脑部和全脊柱再分期MRI未见疾病证据。然而数周后,他出现左髋部疼痛和体重减轻。体能状态下降至ECOG 3级,促使扩大再分期检查范围,包括神经外部位。18-FDG PET-CT显示广泛转移,累及肝脏、骨骼、肾脏、胰腺和淋巴结(右侧锁骨上内侧、纵隔、左侧闭孔、髂外和腹股沟)(图1e)。左股骨颈存在一处FDG高摄取的不规则骨性病变,导致骨折,与左髋部疼痛相符(图4a, b)。对肝脏病灶进行了活检,组织病理学检查显示为低分化肿瘤,由片状和巢状排列的小至中等大小细胞构成,胞浆丰富嗜酸性,核偏位(图5a)。细胞核呈圆形至卵圆形,核膜不规则。可见散在的大型双核和多核细胞,伴有怪异核。免疫组化研究显示肿瘤细胞突触素、CD56、结蛋白阳性,EMA和CD99局灶阳性,MyoD1、肌细胞生成素、波形蛋白、GFAP、角蛋白CAM5.2、SMA、CK7和CK20阴性。Ki67增殖指数为 80–90%。INI-1显示核表达缺失(图5b–f)。治疗两周后,患者临床状况进一步恶化,遂停止治疗。脑部和脊柱MRI显示,与右侧额叶前部相邻的硬脑膜内有一处 2.3×2.1 的对比增强病灶,符合局部复发。中枢神经系统内发现多处新增病灶。一周后,患者因全身疾病进展的临床表现而死亡。总生存期为 11 个月。从发病到复发的时间为 9 个月。复发后生存期为 2 个月。靶向二代测序(NGS)panel(中枢神经系统和肝脏病灶)以及胚系靶向NGS panel的结果见表1。对脑部和肝脏肿瘤病灶进行的比较基因组分析显示,脑部和肝脏病灶中存在两个相同的致癌性/可能致癌性变异:RB1:c.2086_2087insAT(脑部VAF:93%,肝脏VAF:12%)和MPL:c.317C>T(脑部VAF:53%,肝脏VAF:48%)。这两个变异均未在外周血正常细胞中发现。这些结果证实了中枢神经系统和肝脏病灶具有共同的克隆起源。SMARCB1或SMARCA4基因中未发现单核苷酸变异或插入缺失变异。在中枢神经系统病灶中检测到一个潜在的SMARCB1拷贝数变异(chr22: 24,129,273–24,176,467, Hg19);但无法区分是单等位基因还是双等位基因缺失。然而,INI-1表达缺失提示SMARCB1双等位基因失活。在外周血正常细胞中发现了一个致病性胚系TP53缺失变异(NM_000546.5:外显子2–6缺失)(表1)。由于可用组织已耗尽,未能成功进行甲基化分析以确定ATRT分子亚型。基于伦理方面的考虑排除了对患者进行额外取样的可能性。
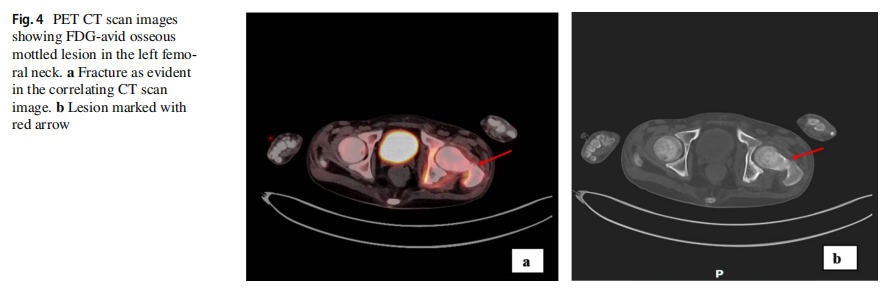
图4 PET-CT影像显示左股骨颈存在FDG摄取性斑点状骨病变
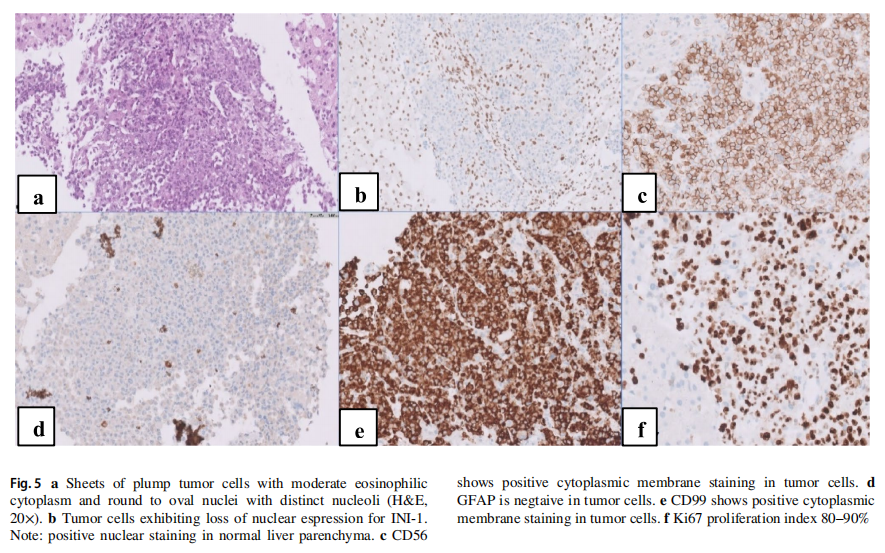
图5 组织学检查结果
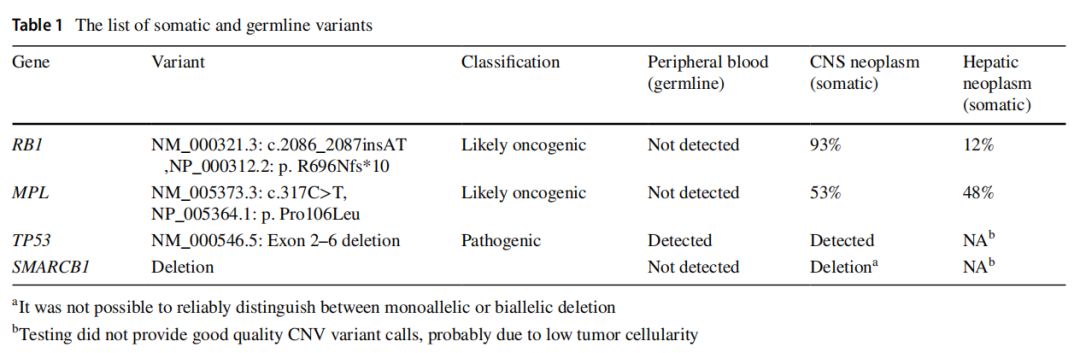
表1 胚系和体系结果
讨 论
本文描述了一例在ATRT达到完全缓解后出现多个进行性生长的神经外横纹肌样病灶的不寻常病例。ATRT的神经外转移极为罕见,文献中仅有少数报道。Bush等人描述了一例ATRT孤立性腋窝淋巴结转移的儿童病例,但未发现其他颅外转移灶或原发肿瘤。Yaguchi等人报告了一例 5 岁女孩的髓外脊柱ATRT病例,伴有脑脊液播散和肺转移。Varan等人记录了一例幕上ATRT患者在诊断后 14.8 个月出现肺转移的病例。相比之下,ATRT与MRT之间的关联并不罕见。因此,在同一患者中区分ATRT的神经外复发与第二原发性MRT可能造成显著的诊断困难。必须考虑多个流行病学、临床和分子层面的论据。
首先,虽然继发性神经外MRT比系统性ATRT播散更为常见,但它们通常发生于携带SMARCB1胚系突变(横纹肌样瘤综合征)的患者,并与分子亚组1("ATRT-MYC样")相关。遗憾的是,研究人员未能通过甲基化分析确认患者的分子亚型。由于记录有限,其家族史尚不明确;然而,胚系分析未发现SMARCB1和SMARCA4基因中存在任何致病性或可能致病性变异。其次,复发发生在治疗第一年内,且神经外病灶先于中枢神经系统复发出现,这种模式更符合原始ATRT的克隆演化,而非新发恶性肿瘤。第三,中枢神经系统和肝脏病灶中共享高变异丰度的致癌性变异(RB1、MPL),且正常组织中不存在,这为共同的克隆起源提供了确凿证据。综上所述,这些发现强烈支持复发的诊断,而非第二原发肿瘤。
对在本文患者中发现的致病性胚系TP53变异的解读则更为复杂。大多数ATRT不携带TP53基因的任何突变。一项对 25 例伴有SMARCB1改变的横纹肌样瘤进行的研究,检测了 115 个致癌基因和抑癌基因的突变情况。所有病例均未发现TP53突变。研究者仅检出一个NRAS激活突变,而所有其他检测的基因中完全不存在致癌性突变。
TP53体细胞变异是多种癌症中最常见的突变之一,但在横纹肌样肿瘤中极为罕见。体外研究表明,完整的MYC-p19ARF-MDM2-p53轴对于SMARCB1缺陷型横纹肌样肿瘤的肿瘤发生至关重要,这可能是这些病例中TP53突变发生率低的原因。然而,TP53是一种多功能蛋白,其变异可通过多种机制促进肿瘤发生。SMARCB1/SMARCA4是SWI/SNF染色质重塑复合体的关键组分,已知在转录调控中也与TP53相互作用。因此,一部分携带胚系TP53变异的ATRT可能源于转录程序的改变,这是合理的。对携带胚系TP53变异的ATRT形态学特征的回顾显示,主要为低分化肿瘤,伴有局灶性横纹肌样分化和可变免疫表型(表2)。这表明携带胚系TP53变异的患者可能代表ATRT的一个独特亚型。
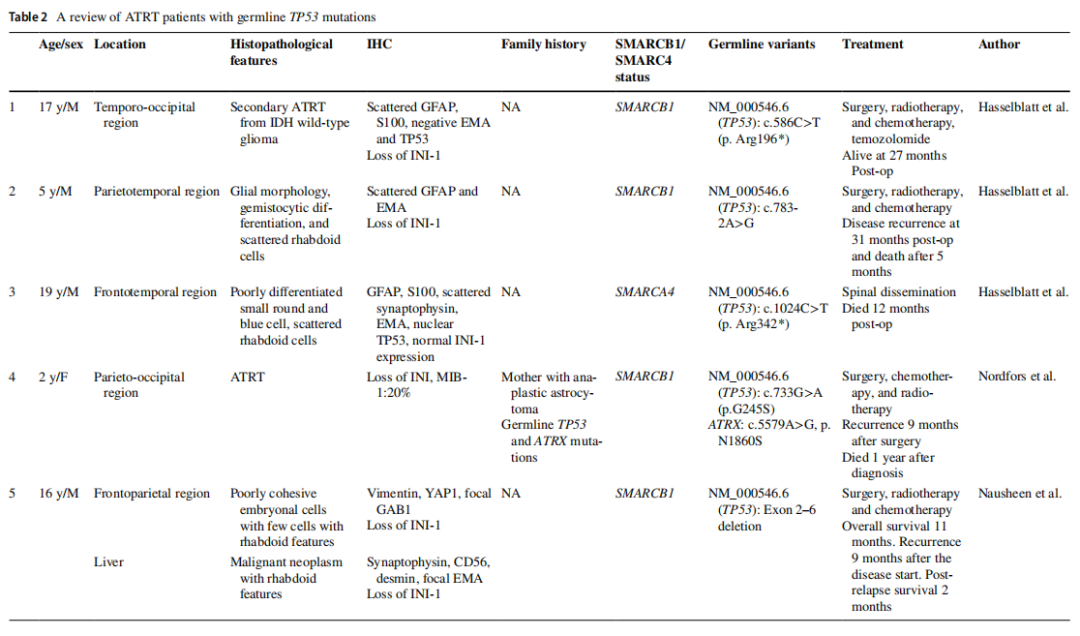
表2 携带胚系TP53突变的ATRT患者综述
ATRT或恶性横纹肌样瘤(MRT)的家族性易感性通常由SMARCB1(罕见情况下为SMARCA4)的胚系功能缺失变异介导,而胚系TP53突变(与李法美尼综合征相关)通常不与ATRT相关。然而,少数文献报告提示存在罕见的例外情况。Nordfors等人描述了一名 5 岁SMARCB1缺陷型ATRT患儿携带胚系TP53突变,其母亲有间变性星形细胞瘤病史。Hasselblatt等人报告了三例额外的SMARCB1或SMARCA4缺陷型ATRT伴胚系TP53变异的病例。有趣的是,Hasselblatt等人报告中的所有三例患者的年龄均"高于预期":分别为 19 岁、17 岁和 5 岁,其中两例为青少年,与本文患者相似。本文病例具有多个与继发性ATRT相似的临床和分子特征,例如诊断年龄高于该肿瘤典型分布的中位年龄,以及存在通常在原发性ATRT中不预期的额外突变。然而,组织病理学检查未发现任何胶质或非横纹肌样成分,而这些成分通常与继发性ATRT相关。
原发性中枢神经系统肿瘤的神经外转移相对少见,报告发生率最高可达 4.3%。颅外播散的危险因素包括手术干预、脑脊液分流术、脑膜受累以及特定的组织学类型,如生殖细胞瘤、髓母细胞瘤和胶质母细胞瘤。已有多种理论被提出来解释脑肿瘤系统性播散的罕见性。这些假说通常侧重于外在屏障(例如,缺乏颅内淋巴管、血脑屏障完整性或脑内微环境)或肿瘤内在特性(例如,突变、细胞运动性或黏附分子表达)。在本文病例中,研究人员推测肿瘤直接侵犯头皮可能通过绕过血脑屏障而促进系统性播散。这一机制可以解释本文患者所观察到的不寻常转移模式。
关于治疗ATRT颅外复发的文献很少。Varan等人报告了一例肺转移性ATRT病例,采用顺铂和依托泊苷治疗;然而,患者在治疗开始后 2.2 个月内因进行性肝转移而死亡。与本文病例相似,含铂化疗未能控制全身性疾病,凸显了神经外ATRT复发的侵袭性。鉴于此类病例极为罕见且预后极差,组织专门的临床试验可能不切实际。考虑到缺乏循证疗法以及顺铂/依托泊苷等常规方案的不良结局,研究人员认为在此情况下,基于分子分析或临床前数据指导的同情性使用超说明书药物在伦理上应是允许的。
在本文病例中,使用多基因panel进行比较突变分析被证明对于区分肿瘤复发与第二原发恶性肿瘤至关重要。此类诊断不确定性在肿瘤学中很常见,尤其是当罕见的转移模式挑战传统范式时。研究人员主张开展前瞻性临床研究,评估基因组分析在解决这些困境中的实用性。尽管极为罕见,但在适当的临床背景下应考虑ATRT的神经外转移。基因检测有助于区分复发与继发性肿瘤。
参考文献:
Yaqoob N, Itkin B, Tajammul SS, Al Jabri MM, Niaz Z, Balaji R, Al Azri A, Alhaddabi IH, Al-Zadjali S, Deshpande P. Atypical teratoid rhabdoid tumor with extraneural metastases: a case report with genomic analysis. Childs Nerv Syst. 2025 Dec 11;41(1):413. doi: 10.1007/s00381-025-06999-8. PMID: 41379345.
小提示:本篇资讯仅在梅斯医学APP中开放阅读,请扫描二维码直接下载APP

猜你喜欢
- 眼睛一直发红流泪,用了很多眼药水都不好?医生一查:竟是“神经营养性角膜炎”在作怪!
- 吃豆腐的好处 榨菜豆腐汤的功效
- 古诗云春眠不觉晓 3招赶走春困让你精神饱满
- 来曲唑对生育力保存患者促排卵过程和妊娠结局的影响
- 2022年2月18日简报:香港疫情日增超6000,以BA.2为主;73%美国人已对奥密克戎免疫;意大利新冠死者96%为退休老人,一年减少11亿欧元养老金
- 秋季人手凉该吃什么好呢?
- 上班族肩膀酸痛按摩有用吗
- 葱爆河虾的做法-酸辣味其他工艺菜谱
- 解析疾病“信使”探究生命之谜(大健康观察)
- 【愈见乙肝2023】一对HBV DNA、HBsAg和HBeAg高水平的母女经个体化抗病毒治疗均获长期持久的临床治愈